آموزش نرم افزار VMD برای شبیه سازی دینامیک مولکولی – به زبان ساده
شبیهسازی دینامیک مولکولی روشی برای بررسی حرکت فیزیکی اتمها و مولکولها است. در این روش حرکت اتمها و مولکولها در یک بازه زمانی مشخص بررسی و برای محاسبه مقدار عددی حرکت مولکولها در واحد زمان از معادله حرکت نیوتن استفاده میشود. در این معادله از پتانسیل بین اتمی و «میدان نیروی مکانیکی مولکول» (Molecular Mechanical Force Fields) برای محاسبه نیروی بین مولکولها و انرژی پتانسیل آنها استفاده می شود. این روش در شیمیفیزیک، مهندسی مواد، بیوفیزیک، برهمکنش پروتئینها، برهمکنش مولکولها با غشای لیپیدی و طراحی سیستمهای دارویی جدید کاربرد دارد. نرم افزار VMD یکی از نرمافزارهای متداولی است که برای نمایش تصویری محاسبات شبیهسازی دینامیک مولکولی از آن استفاده میشود. به همین دلیل آموزش نرم افزار VMD برای افرادی که از شبیهسازی دینامیک مولکولی استفاده میکنند از اهمیت ویژهای برخوردار است.
- یاد میگیرید ساختار و عملکرد کلیدی نرمافزار VMD را شرح دهید.
- نحوه بارگذاری و مدیریت انواع فایلهای مولکولی را میآموزید.
- خواهید آموخت چگونه ظاهر و نمایش مولکولها را سفارشی کنید.
- با روشهای آنالیز دادههای دینامیک و اجرای اسکریپت آشنا میشوید.
- یاد میگیرید سرعت انیمیشن و فریمها را تنظیم و کنترل کنید.
- خواهید توانست جعبه PBC را رسم و شخصیسازی کنید.




این نرمافزار با دریافت فایل مختصات اتمها، توپولوژی و تغییرات مختصات در واحد زمان (فایل تراجکتوری) ساختار مولکول یا سیستم شبیهسازی شده را نمایش میدهد. این نرمافزار از زبانهای Tclو پایتون پشتیبانی میکند. به همین دلیل کاربر میتواند علاوه بر استفاده از کلیدهای گرافیکی این نرمافزار از کدهای دستوری دلخواه برای رسم مولکول و انجام آنالیزها استفاده کند. در مطلب آموزش نرم افزار VMD مجله فرادرس، شیوه نمایش مولکولها در نرمافزار VMD را به همراه تعدادی از آنالیزهای مولکولی این نرمافزار توضیح میدهیم.
نرم افزار VMD چیست؟
VMD نرمافزاری است که برای مدلسازی، تصویرسازی و آنالیز مولکولهای زیستی ازجمله پروتئینها، نوکلئوئیکاسیدها، غشای دو لایه، میسل و لیپوزومها طراحی شده است. این نرمافزار به کاربر کمک میکند، فایل PDB پروتئینها و ساختار نهایی مولکول شبیهسازی شده در شبیهسازی دینامیک مولکولی را با انواع ساختارهای مولکولی استاندار مشاهده کند. این نرمافزار محدودیتی برای نشان دادن تعداد اتمهای مولکول ندارد. این سیستم قابلیت نمایش فایل تراجکتوری شبیهسازی AMBER، Charmm، Gromacs، MMTK، NAMD، X-Plor به شکل انیمیشن را دارد. به کمک این قابلیت میتوان روند تشکیل ساختار یا تغییرات زاویهها، فاصله بین اتمها و تغییر انرژی در طول زمانرا بررسی کرد. به همین دلیل آموزش نرم افزار VMD یکی از ضروریات انجام شبیهسازیهای دینامیک مولکولی است.
این نرمافزار قابلیت به تصویر کشیدن و تولید فایل مختصات مولکول با فرمت CryoEM، پتانسیل الکترواستاتیک و چگالی الکترون را دارد. نرمافزار VMD امکان آنالیز متغیرهای مختلف مولکول و اتمهای آن ازجمله مرکز جرم و شعاع ژیراسیون را فراهم میکند و قابلیت خواندن دستورات به زبان پایتون و Tcl را دارد. به همین دلیل میتوان به جای استفاده از کلیدهای گرافیکی این نرمافزار از کدهای دستوری برای تغییر دادن مولکول استفاده کرد. این نرمافزار در سه نسخه مخصوص Windows، MacOS و Unix در سایت رسمی نرمافزار وجود دارد. آموزش نرم افزار VMD در این مطلب را با توضیح بخشهای مختلف نوار کاربری شروع میکنیم و در انتها شیوه خروجی گرفتن از این نرمافزار را توضیح می دهیم.
- لینک وبسایت نرمافزار VMD: «+»
نوار کاربری نرم افزار VMD
اولین قدم آموزش نرم افزار VMD آشنایی با نوار کاربری این نرمافزار است. برای باز کردن نرمافزار VMD در سیستم عاملهای ویندوز و مک روی آیکون نرمافزار کلیک و برای باز کردن نرمافزار در سیستم عامل Unix کلمه VMD را در ترمینال تایپ کنید. دو پنچره برای شما باز میشود. پنجره اصلی (سفید) برای تغییر پارامترهای نمایش و آنالیز مولکولها، و پنجره گرافیکی (سیاه) برای نمایش شکل مولکول است. بخش بالایی نوار کاربری شامل گزینههای File و Molecule و Graphics و Display و Mouse و Extensions و Help است و نوار پایینی بخشی برای تنظیم سرعت نمایش فایل تراجکتوری است. مشخصات فایل باز شده در کادر بین این دو نوار نمایش داده میشود.
گزینه File نوار کاربری نرم افزار VMD
با استفاده از آیتم "New Molecule" از گزینه "File" میتوانید فایل مولکول جدید را باز کنید. برای مشاهده ساختار پروتئینها میتوانید فایل متن PDB را از سایت دانلود و با کلیک روی "Browser" کادر "File Name" در پنجره باز شده، پروتئین را در VMD بارگذاری کنید. با کلیک روی "Load" ساختار پروتئین در پنجره گرافیکی نمایش داده میشود. فایل توپولوژی و تراجکتوری مولکولها را میتوان با همین روش در VMD بارگذاری کرد. قبل از لود فایل تراجکتوری حتما باید فایل توپولوژی مولکول در سیستم بارگذاری شود. به کمک آیتمهای گزینه File میتوانید دستورات زبان Tcl را در کنسول نرمافزار یا فایل مولکول اعمال و فایل خروجی را دریافت کنید.
پس از بارگذاری فایل مختصات مولکول یا تراجکتوری شبیهسازی ویژگیهای آن در صفحه اصلی نمایش داده میشود. "ID" نامی است که نرم افزار VMD برای مولکول انتخاب میکند. "Molecule" نام فایل توپولوژی مولکول در سیستم کاربر است. Atoms تعداد اتمها و Frames تعداد فریمهای مولکول را نشان میدهد. Top | T و Active| A و Drawn| D و Fixed | F سمت چپ نام فایل مولکول نمایش داده میشوند و وضعیت مولکول را نشان میدهند.

- Top: حرف T کنار نام مولکول نشانه اعمال شدن تغییرات گرافیکی و دستورات روی این مولکول است.
- Active: حرف A کنار نام مولکول به معنی فعال بودن مولکول است. مولکولهای فعال در انیمیشن شبیهسازی ثابت نیستند و دستورات VMD روی آنها اعمال میشود.
- Drawn: حرف D کنار نام مولکول به این معنی است که مولکول در پنجره گرافیکی نمایش داده نمیشود و به طور موقت حذف شده است.
- Fixed: حرف F کنار نام مولکول به این معنی است که مولکول در حالت ثابت نه دینامیک قرار دارد. به این نکته توجه کنید که مختصات اتمهای مولکول با تغییر زاویه نمایش آن بهوسیله ماوس، تغییر نمیکند و موقعیت اتم ثابت است.
گزینه Graphics نوار کاربری نرم افزار VMD
در این بخش از آموزش نرم افزار VMD نحوه تغییر ویژگیهای گرافیکی مولکول را توضیح میدهیم. Representation و Colors و Materials و Labels و Tools آیتمهای گزینه "Graphics" هستند که نحوه نمایش، ساختار و رنگ مولکول را مشخص میکنند.

تغییر ساختار مولکول در نرم افزار VMD با گزینه Representation نوار ابزار
تغییر ساختار مولکول یکی از مطالب مورد توجه افرادی است که آموزش نرم افزار VMD را دنبال میکنند. برای تغییر ساختار میتوانید از آیتم "representation" استفاده کنید. نوار بالایی پنجره representation شامل گزینههای "Selected Molecule" و "Create Rep"، "Delete Rep" و بخش پایینی آن شامل تبهای "Draw Style"، "Selection"، "Trajectory"، و "Periodic" میشود.
- Selected Molecule: در این کادر نام مولکول انتخاب شده نمایش داده میشود. کاربر بهوسیله نوار کشویی این بخش میتواند مولکول را تغییر دهد.
- Create Rep: کاربر با استفاده از این گزینه میتواند مولکول جدید به مولکول قبلی اضافه و هر دو را در یک صفحه نمایش مشاهده کند.
- Delete Rep: با استفاده از این گزینه میتوان یکی از مولکولها را از صفحه نمایش حذف کرد. برای این کار مولکول مورد نظر را در کادر "Selected Molecule" انتخاب و روی گزینه "Delete Rep" کلیک کنید. به جای حذف مولکول میتوانید مولکول به طور موقت حذف کنید. برای این کار دو بار روی نام مولکول در کادر "Selected Molecule" کلیک کنید. نام مولکول به صورتی تغییر میکند و مولکول پنهان میشود.
- Draw Style: کادر "Drawing Method" در این تب مشخص میکند مولکول با چه ساختاری نمایش داده شود. "Coloring Method" رنگ هر اتم و "Materials" ویژگیهای بصری (نور، سایهروشن رنگها و شفافیت) مولکول را مشخص میکند. به کمک "Coloring Method" کاربر انتخاب میکند رنگ تمام بخشهای مولکول را همزمان یا بر اساس بعضی از ویژگیها ازجمله ساختار دوم، بار الکتریکی، جرم مولکولی، نوع باقیماندههای آمینواسیدی (بازی یا اسیدی) و کنفورماسیون تغییر دهد.
- Selection: کاربر در کادر "Singlewords" میتواند گروهی از مولکولها یا اتمها را برای نمایش انتخاب کند. با انتخاب گزینه all تمام اتمها به نمایش گذاشته میشود و با انتخاب none تمام اتمها از صفحه نمایش حذف میشوند. با انتخاب یکی از گزینههای and، or و not در کنار این کادر، میتوان مولکولها یا بخشی از مولکول را همزمان یا جدا نمایش داد. بهوسیله آيتم "Value" تغییرات را روی یک نوع از اتمها اعمال کرد.
- Trajectory: کاربر با استفاده از این گزینه میتواند سرعت حرکت مولکول انتخابی در بخش selection را در انیمیشن فایل تراجکتوری تغییر دهد. افزایش عدد کادر "Trajectory Smoothing Window" در این تب، سرعت حرکت مولکول یا بخش انتخاب شده از آن را کاهش و کاهش آن سرعت حرکت را افزایش میدهد.
- Periodic: آیتمهای این گزینه تعداد جعبههای PBC (شرایط مرزی متناوب | Periodic Boundary Condition) را تغییر میدهد. با انتخاب گزینه X+ یک جعبه در جهت مثبت محور X کنار جعبه مرکزی ایجاد میشود. کادر "Number of Images" تعداد جعبههای ایجاد شده در ۶ جهت فضای اطراف جعبه مرکزی را تعیین میکند.

تغییر رنگ در VMD با گزینه Color نوار ابزار
در این بخش از آموزش نرم افزار VMD تغییر رنگ مولکولها و نمودارها در نرمافزار را توضیح میدهیم. آیتم Colors این امکان را در اختیار کاربر میگذارد که رنگ اتمها، نمودارها، پسزمینه پنجره گرافیکی و برچسبهای ایجاد شده را تغییر دهد. در این نرمافزار ۱۶ رنگ پیشفرض وجود دارد و کاربر میتواند رنگ جدیدی به آنها اضافه کند.

Materials
آیتمهای این گزینه ویژگیهای بصری مولکول را تغییر میدهد. بالا سمت چپ پنجره این گزینه تمام ویژگیهایی که قبلا تنظیم شده نشان داده میشود. زیر این کادر پنج کادر وجود دارد که تغییر اسلاید آن، ویژگیهای مولکول را تغییر میدهد. کاربر با کلیک روی کلید "Create New" بالا و راست این پنجره میتواند تم جدیدی برای نمایش مولکولها ایجاد کند و با کلیک روی گزینه "Default" تغییرات ایجاد شده به حالت پیفرض نرمافزار برمیگردد.
Labels
آیتمهای این پنجره برچسبهای اتمها را تغییر میدهد. از نوار کشویی بالا سمت چپ پنجره این گزینه میتوان برچسبهای Atoms، Bonds، Angles، Dihedrals و Springs را انتخاب کرد. با انتخاب Atoms نام اتمها در شکل نشان داده میشود. با انتخاب گزینه Bonds پیوند بین دو اتم با خطچین نشان داده میشود و اندازه آن برحسب آنگستروم وسط خط نوشته میشود. Angles شکل زاویه تشکیل شده بین سه اتم را با خطچین و مقدار عددی آن را با درجه نمایش میدهد. Dihedrals زاویه چرخشی ایجاد شده بین چهار اتم (زاویه بین صفحه تشکیل شده از سه اتم اول و صفحه تشکیل شده از سه اتم آخر) و مقدار عددی آن را نشان میدهد. Springs فاصله بین هر دو اتم را با خطچین و برحسب آنگستروم نمایش میدهد.
سه گزینه سمت راست سمت کادر انتخاب برچسب وجود دراد. گزینه "Delete" برچسب انتخابی را حذف میکند. با انتخاب گزینه "Hide" برچسب به طور موقت نشان داده نمیشود و با انتخاب گزینه "Show" نشان داده میشود. "Pick Atom" اولین تب پایین پنجره Labels است که مختصات و ویژگیهای آخرین اتم انتخاب شده را نشان میدهد. مقادیر عددی این برچسبها در طول زمان را میتوان با استفاده از تب "Graph" به نمودار تبدیل کرد.
گزینه Display نوار کاربری vmd
گزینه Display ویژگیهای مولکول در پنجره گرافیکی را تغییر میدهد. Reset View و Stop Rotation و Perspective و Orthografic و Depth Cueing و Culling و FPS و Lights و Axes و Background و Stage و Sterio و Chachemode و Render Mode و Antialiasing و Display Setting آیتمهایی هستند که در این بخش از مطلب مجله فرادرس کاربرد آنها را توضیح میدهیم.
- Reset View: به کمک این آيتم میتواند نیروی اعمال شده به مولکول را حذف و مولکول را به مقیاس اولیه برگرداند.
- Stop Rotation: این گزینه چرخش خودکار مولکول در صفحه نمایش را متوقف میکند. چرخش خودکار با حرکت سریع ماوس همراه فشار لحظهای کلید شروع میشود. برای توقف این حالت میتوان از فشار دکمههای ماوس هم استفاده کرد.

- Perspective: مولکول بدر دو حالت Perspective و Orthografic در پنجره گرافیکی نمایش داده میشود. در نمای Perspective اتمهای دوتر کوچکتر از اتمهای نزدیکتر هستند. در نمای Orthografic اتمهای دور و نزدیک به یک اندازه نمایش داده میشوند.
- Antialiasing: به کمک این گزینه میتوانید لبههای ناصاف ایجاد شده به دلیل رزولوشن و پیکسلهای صفحه نمایش در ساختار مولکول را تغییر دهید. این گزینه دو آيتم on و off دارد.
- Depth Cueing: این گزینه دو آیتم on و off دارد. در حالت on اتمهای دورتر مولکول با رنگ زمینه ادغام میشود و درک بهتری از عمق سهبعدی مولکول ایجاد میکند. کاربر میتواند آيتمهای این گزینه را در Display Setting تغییر دهد. پارامتر Cue Mode معادله محو کردن مولکول را تعیین میکند. در حالت "Linear" شیب یکنواختی از محو شدن با نقطه شروع و پایان مشخص ایجاد میکند. Exp و Exp2 بر اساس تراکم عمل و مولکول را بهتر محو میکنند.
- Culling: این گزینه مثل دو گزینه قبلی دو آیتم on و off دارد. این گزینه کیفیت تصوریر خروجی را افزایش میدهد.
- FPS: این گزینه در حالت enable سرعت خروجی تصویر یا فیلم مولکول را گوشه بالا و راست پنجره گرافیکی نشان میدهد.
- Lights: آيتمهای این گزینه نور صفحه نمایش را تغییر میدهد.
- Axes: کاربر بهوسیله این گزینه میتواند کنسول محورهای X، Y و Z را در یکی از چهار گوشه یا مرکز صفحه نمایش قرار دهد یا حذف کند.
- Background: آیتمهای این گزینه پسزمینه صفحه نمایش را کامل یا در قالب پررنگ به کمرنگ (از بالا به پایین) تغییر دهد.
- Stage: تعداد مراحل شبیهسازی را در یکی از چهار گوشه یا مرکز صفحه نمایش نشان میدهد؟ و میتوان آن را حذف کرد.
- Cachemode: آیتمهای این گزینه مشخص میکند، سیستم از آرشیو تصویری برای افزایش سرعت خروجی گرفتن استفاده کند.
- Rendermode: آیتمهای این گزینه نحوه خروجی گرفتن را بر اساس کارت گرافیک سیستم تنظیم میکند.

گزینه Mouse نوار کاربری نرم افزار VMD
اگر تا این بخش از آموزش نرم افزار VMD ما را همراهی کرده باشید، با لود کردن فایلها و تغییر ساختار مولکول در این نرمافزار آشنا شدهاید. در این بخش از مطلب مجله فرادرس آیتمهای گزینه "Mouse" نوار کاربری را توضیح میدهیم. گزینه Mouse حرکت ماوس در پنجره گرافیکی را کنترل میکند. حرکت ماوس در پنجره گرافیکی وضعیت مولکول را تغییر میدهد و برای کاربر این امکان انتخاب کردن یک یا چند مولکول را فراهم میکند. "Mouse Mode" و "Pick Mode" دو بخش این گزینه هستند. "Rotate"، "Translate" و "Scale" سه آیتم اول این گزینه هستند که ماوس را در حابت ترجمه، مقیاس و چرخش قرار میدهند. اگر گزینه "Rotate" را انتخاب کنید، با کلیک دکمه چپ و حرکت دادن ماوس، موقعیت فضایی مولکول در پنجره گرافیکی تغییر میکند.
با انتخاب گزینه "Translate"، فشار دادن کلید چپ و حرکت دادن ماوس میتوانید مولکول را در بخشهای مختلف پنجره گرافیکی حرکت دهید. پس از انتخاب گزینه "Scale"، فشار دادن کلید چپ و حرکت دادن ماوس در جهت افقی میتوانید بزرگنمایی مولکول را افزایش یا کاهش دهید. Center و Query و Label و Move و Force و Move Light و Add/Remove Bonds گزینههای دیگر این بخش از نوار کاربری هستند. که اثر ماوس روی مولکول را تغییر میدهند.
- Center: با انتخاب این گزینه اگر نشانه ماوس را روی یک اتم قرار دهید، روی دکمه چپ کلیک کنید و ماوس را حرکت دهید، کل مولکول حول اتم انتخاب شده میچرخد. اگر در این حالت گزینه "Scale Mode" را انتخاب کنید، صفحه نمایش مولکول بزرگتر میشود و همچنان اتم انتخاب شده مرکز مولکول است. اتم انتخاب شده تا زمانی که اتم دیگری را انتخاب یا مولکول دیگری را لود کنید یا گزینه "Reset View" را انتخاب کنید مرکز مولکول باقی میماند.
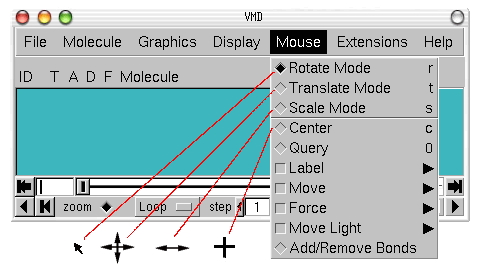
فلشهای پایین نحوه نمایش ماوس در پنجره گرافیکی پس از انتخاب گزینهها را نشان میدهد. - Query: با انتخاب این گزینه پس از کلیک روی هر اتم، اطلاعات آن (برای مثال نام اتم) در کنسول متن نشان داده میشود.
- Label: با انتخاب این گزینه به مولکول برچسبهای ازجمله نام اتم، طول پیوندها، زاویه بین پیوندها، زوایای دیهیدرال را اضافه میکنیم. برای اضافه کردن این برچسبها به ترتیب انتخاب یک، دو، سه و چهار اتم نیاز است. مقادیر عددی پیوند، زوایای بین پیوندی و زوایای دیهیدرال در برچسبها نمایش داده میشود و دو اتم انتخاب بهوسیله خطچین به هم وصل میشود. به علاوه میتوانید آیتمهای "Label" را از پنجره "Label" گزینه "Graphics" نوار کاربری تغییر دهید.


فاصله دو اتم انتخابی با خطچین و فاصله بین اتمها برحسب آنگستروم نمایش داده شده است.
- Move: آیتمهای این گزینه مختصات اتمهای در صفحه نمایش را تغییر میدهد. با انتخاب "Atom" از نوار کشویی این گزینه فقط اتمهای اننتخاب شده حرکت و مختصات آنها تغییر میکند. با انتخاب "Residue" تمام اتمهای مشابه در باقیماندههای مشابه برای مثال (باقیماندههای آمینواسیدی یا نوکلئوتیدی) را تغییر میدهد. انتخاب گزینه "Fragment" مختصات تمام اتمهای متصل به اتم انتخابی را تغییر میدهد. انتخاب "Molecule" تمام اتمهای موجود در مولکول را حرکت میدهد و "Highlight Rep" مختصات تمام اتمهای انتخابی در پنچره "Graphics" را تغییر میدهد. اگر با نگهداشتن همزمان کلید "Shift" کیبورد و دکمه چپ ماوس، ماوس را حرکت دهید، اتمها حول اتم انتخاب شده میچرخند. اگر با نگه داشتن کلید چپ، ماوس را حرکت دهید، مختصات اتمها در محور X یا Y صفحه نمایش تغییر میکند و اگر با نگه داشتن دکمه راست یا میانی ماوس را حرکت دهید، اتمها در محور عمود بر صفحه نمایش حرکت میکنند. این گزینه غیرقابل برگشت است. به همین دلیل قبل از تغییر مختصات یک کپی از فایل اصلی تهیه کنید.
- Force: آیتمهای این گزینه به کاربر کمک میکند اثر نیرو به اتمها، باقیمانده یا قطعهای مولکول را مشاهده کند. این گزینه در شبیهسازی دینامیک برهمکنش مولکولی (Interactive Molecular Dynamics | IMD) کاربرد دارد. فشار دادن کلید چپ و حرکت ماوس به اتم انتخابی نیرو وارد و فشار دادن کلید راست یا میانی نیرو را حذف میکند.
- Move Light: آيتمهای این گزینه نور اطراف مولکول را تغییر میدهد.
- Add/Remove Bonds: بهوسیله این گزینه میتوانید بین اتمهای یک مولکول پیوند جدید ایجاد یا پیوندهای قبلی را حذف کنید.
گزینه Extensions نوار کاربری نرم اافزار VMD
با استفاده از آيتم Analysis گزینه Extensions نوار ابزار پنجره اصلی میتوان مولکول را آنالیز کرد. آیتم TK Console این گزینه امکان اعمال کدهای دستوری و آیتم Simulation این بخش از نوار کاربری امکان شبیهسازی دینامیک برهمکنش مولکولی را برای کاربر فراهم میکند.
آنالیز مولکول در نرم افزار VMD
آنالیز مولکولها یکی از بخشهای مهم آموزش نرم افزار VMD است. VMD یکی از نرمافزارهای کاربردی در آنالیز ساختار و مراحل شبیهسازی مولکولها است. علاوه بر گزینههای آنالیز خود نرمافزار کاربر میتواند با استفاده از دستورات Tcl در کنسول TK آنالیزهای دلخواه خود را انجام دهد. تغییر فاصله مولکولها نسبت به زمان در فایل تراجکتوری یکی از پارامترهایی است که میتوان بهوسیله این نرمافزار اندازه گرفت.
برای این کار از گزینه "Mouse" آیتم "Label" و "Bonds" را انتخاب کنید. با کلیک ماوس روی اتمهای مورد نطر فاصله بین آنها برحسب آنگستروم در صفحه گرافیکی نمایش داده میشود. از گزینه "Graphics" آیتم "Graph" را انتخاب و روی پیوندی که در پنجره گرافیکی مشخص کردهاید، کلیک کنید. در این حالت نمودار تغییر فاصله دو اتم نشان داده میشود.
سطح در تماس مولکول با حلال (SASA) یکی دیگر از ویژگیهایی است که میتوان با VMD محاسبه کرد. برای این آنالیز فابل تراجکتوری مولکول را در نرمافزار لود کنید. تصویر زیر دستورات Tclبرای محاسبه SASA را نشان میدهد. این دستورات را در "Notepad" سیستم ذخیره کنید.

از گزینه "Extensions" کنسول TK را انتخاب کنید. کلمه source و نام فایل در کنسول تایپ کنید. نام مولکول را روبروی selection وارد کنید. نرمافزار آنالیز SASA را شروع میکند.

فایل خروجی SASA مولکول در هر فریم را نشان میدهد.

محاسبه شعاع ژیراسیون یکی دیگر از آنالیزهایی است که در مطلب آموزش نرم افزار VMD توضیح میدهیم. این کمیت پراکندگی اتمها نسبت به مرکز جرم مولکول را مشخص میکند. به کمک این پارامتر پایداری مولکول در محلول و میزان فشردگی آن را تعیین میکنیم. به علاوه در بعضی سیستمها ازجمله سیستمهای لیپوزومی به کمک این پارامتر میتوان شکل سیستم (برای مثال لیپوزوم بیضی یا کروی) را مشخص کرد. برای محاسبه شعاع ژیراسیون به سه فایل دستور نیاز دارید. این دستورات در بخش "Script Library" سایت اصلی این نرمافزار وجود دارد. دستور اول مرکز جرم مولکول را محاسبه میکند.

در فایل دوم دستورات محاسبه شعاع ژیراسیون نوشته شده است.

در فایل سوم دستورات لوپ تکرار برای تمام فریمها نوشته شده است.

برای شروع آنالیز، کنسول Tk را باز و نام فایل سوم (در این مثال rog_loop_dcd.tcl) را پس از کلمه source تایپ کنید. کلید "Enter" تا محاسبات شروع شود. محاسبات بر اساس نوع سیستم کاربر و تعداد فریمها ممکن است از چند دقیقه تا چند ساعت زمان ببرد. فایل خروجی شعاع ژیراسیون مولکول در هر فریم را نشان میدهد.

پل نمکی ترکیبی از نیروی الکترواستاتیک و پیوند هیدروژنی بین تو مولکول است. پارامتر دیگری است که با نرم افزار VMD محاسبه میشود. برای تعیین این پارامتر ابتدا فایل تراجکتوری مولکول مورد نظر را در VMD باز کنید. سپس از پنجره اصلی گزینه Extensions و از نوار کشویی باز شده Analysis را انتخاب کنید. سپس روی آیتم "Salt Bridge" کلیک کنید. از پنجره باز شده تعداد فریمها برای انجام آنالیز، فاصله بین اتمها و محل ذخیره فایل خروجی در سیستم را تعیین کنید. با کلید روی "Find Salt Bridge" محاسبه پل نمکی در فاصله انتخاب شده شروع میشود. فایل خروجی فاصله بین مولکولهای درگیر در پل نمکی را نشان میدهد.
«ریشه میانگین توان دوم انحراف» (Root Mean Square Deviation | RMSD) یکی دیگر از پارامترهایی است که میتوان با نرمافزار VMD محاسبه کرد. این پارامتر میانگین انحراف فاصله دو اتم در پروتئینهای مشابه ار نشان میدهد. هر چه این فاصله کمتر باشد، ساختار دو پروتئین شباهت بیشتری به هم دارد. به کمک این پارامتر میتوان حرکت اتمها در فریمهای مختلف فایل تراجکتوری شبیهسازی مولکولی را محاسبه کرد. برای محاسبه این پارامتر بعد از باز کردن فایل تراجکتوری از منوی پنجره اصلی گزینه Extension و سپس آیتم "RMSD Trajectory Tool" را انتخاب کنید. در کادر بالایی پنجره باز شده نام مولکولی که RMSD آن را محاسبه میکند، وارد کنید. در سمت راست پنجره نام فایل خروجی نوشته شده است. با انتخاب گزینه "Plot" نرمافزار نموداری از تغییر فاصله مولکولها در فریم رسم میکند. انتخاب گزینه align قبل از شروع محاسبات مختصات اتمها در فریمهای فایل تراجکتوری را در یک راستا میکند. با کلیک بر گزینه RMSD محاسبه این پارامتر در نرمافزار انجام و فایل خروجی در سیستم کاربر ذخیره میشود.
نحوه رسم جعبه PBC در نرم افزار VMD چگونه است؟
اگر محدوده مشخصی برای حرکت مولکولها در شبیهسازیهای دینامیک مولکولی مشخص نکنیم، مولکولها تا فاصله بینهایت از هم دور میشوند و ممکن است ساختار مورد نظر تشکیل نشود. جعبه pbc محدوده شبیهسازی و حرکت مولکولها را مشخص میکند. کاربر میتواند برای رسم جعبه pbc از گزینه "Extensions " پنجره "TK console" را باز و pbc box را تایپ کند. با نوشتن این دستور جعبه pbc در پنچره گرافیکی نمایش داده میشود. با انتخاب یکی از گزینههای "Perspective" و "Orthografic" از آیتم "Graphical Representative"شکل جعبه تغییری میکند. برای انتخاب رنگ جعبه میتوانید دستور "color blue -" را به دستور ایجاد جعبه در کنسول اضافه و به جای "blue" از رنگهای دیگر استفاده کنید.

Help
با انتخاب این گزینه یک صفحه وب باز میشود که راهنمای استفاده از نرمافزار و سوالات پر تکرار کاربران در آن وجود دارد.
نوار تغییر سرعت انیمیشن در نرم افزار VMD
در بخشهای قبلی آموزش نرم افزار VMD نحوه لود کردن فایل در نرمافزار، تغییر رنگ و بخشی از آنالیزهای مولکولی را توضیح دادیم. اما چگونه میتوان خروجی دیداری از این نرمافزار دریافت کرد. این ندر این نوار سرعت نمایش انیمیشن مراحل تشکیل مولکول را میتوان به وسیله گزینههای Step و Speed تنظیم کرد. پیشفرض step برای همه مولکولها (۱) است. این عدد نشان میدهد که تمام فریمهای تشکیل مولکول نمایش داده میشود. اگر این عدد را به (۵) تغییر دهید، سرعت انیمیشن بیشتر میشود. چون در این حالت نرمافزار فریمهای مضرب (۵) را نمایش میدهد. پیش فرض speed همیشه در انتهای راست کادر است. تغییر این گزینه به سمت چپ کمترین زمان لازم برای نمایش دو فریم پشت سر هم را افزایش و سرعت انیمیشن را افزایش میدهد.

در نوار بالایی میتوانید فریم مورد نظر خود را مشخص کنید. توجه داشته باشید که شمارش فریمها در این بخش از (0) شروع میشود و برای مشاهده فریم پنجم باید (0) را به (4) تغییر دهید. وقتی انیمیشن تمام میشود بر اساس گزینهای که انتخاب کردید، سه وضعیت ایجاد میشود. اگر گزینه Loop را انتخاب کرده باشید، انیمیشن دوباره از فریم اول پخش میشود. اگر گزینه Once را انتخاب کرده باشید، پخش انیمیشن متوقف میشود و اگر گزینه Rock را انتخاب کرده باشید، انیمیشن از فریم آخر به اول پخش میشود.
نحوه دریافت خروجی تصویر و انیمیشن در نرم افزار VMD چگونه است؟
فایل خروجی نرم افزار VMD را میتوان به صورت عکس، انیمیشن و فایل VMD ذخیره کرد. برای دریافت خروجی تصویر میتوانید از گزینه File نوار کاربری آیتم "Render" را انتخاب کنید. پس از انتخاب این گزینه پنجرهای با سه کادر Render Using، Filename و Render Command برای کاربر باز میشود. گزینه "Filename" نام فایلی که از خروجی گرفته میشود را نشان میدهد.
با انتخاب گزینه "Snapshot" از نوار کشویی "Render Using" نرمافزار از پنجره گرافیکی عکس میگیرد. کاربر میتواند در کادر "Render Command" رزولوشن عکس را مشخص کند. برای مثال اضافه کردن دستور "res 400 300-" تصویری با ابعاد ۴۰۰ در ۳۰۰ پیکسل ایجاد میکند. با کلیک بر دکمه "Start Rendering" فرایند تشکیل تصویر شروع میشود.

کاربر میتواند با انتخاب گزینه "Tachyon" از نوار کشویی "Render Using" تصویر خروجی کیفیت لازم برای تهیه پوستر و چپ در مقالات را دارد. برای ایجاد تصویری با وضوح بیشتر، بهتر از تنظمیات "Shadows" و "Amb. Occl" در "Display Setting" را روی گزینه on و "Material" در "Graphical rep" را روی یکی از گزینههای "AOShiny"، "AOChalchy" یا "AOEdgy" تنظیم کنید.
استفاده از گزینه "POV-Ray 3.6" یکی دیگر از روشهای متداول ایجاد خروجی عکس در نرم افزار VMD است. برای استفاده از این گزینه، نرمافزار "POV-Ray" باید از قبل روی سیستم کاربر نصب شده باشد. این نرمافزار را میتوان رایگان از سایت رسمی آن دریافت کرد. برای نامگذاری تصویر خروجی این روش باید از پسوند "pov." استفاده شود. قبل از شروع دریافت خروجی مطمئن شوید فایل "pvengine.exe" در همان آدرسی که "Render Command" نشان میدهد قرار دارد. در غیر این صورت آدرس اصلی فایل را پیدا و با کلید "Ctri+V" به شکل "آدرس فایل" در کادر کپی کنید.
بعد از دریافت فایل "pov." از VMD این فایل را در نرمافزار "POV-Ray" باز کنید. اگر رزولوشن مورد نظر شما در حالتهای پیشفرض این نرمافزار نبود کافی است از گزینه "Tools" آیتم "Edit resolution INI file" نوار ابزار سایز مورد نظر خود مثل نمونههای قبلی اضافه کنید. سپس از گزینه "File" نوار کاربری آیتم "Open File" را انتخاب و فایل مورد نظر را لود کنید. در نهایت تصویر با کلیک روش گزینه "Run" در نوار کاربری ایجاد میشود.

رنگ و وضوح تصویر ایجاد شده با گزینه "Tachyon" از دو تصویر دیگر بیشتر، اما زمان بیشتری برای دریافت خروجی آن نیاز است.
نرمافزار VMD از هر فریم شبیهسازی دینامیک مولکولی snapshot میگیرد و از ادغام این تصاویر انیمیشن مراحل شبیهسازی و برهمکنش مولکولها را میسازد. برای دریافت خروجی انیمیشن دینامیک از گزینه "Extension" در نوار کاربری آیتم "Visualization" و سپس آیتم "Movie Maker" را انتخاب کنید. در پنجره ایجاد شده میتوانید نام فایل، زاویه چرخش مولکول، شماره stage و زمان انیمیشن را تغییر دهید. برای دریافت خروجی انیمیشن ابتدا فایل تراجکتوری شبیهسازی را در VMD لود کنید. قبل از دریافت خروجی میتوانید سرعت انیمیشن را با روشی که در بخشهای قبلی این مطلب از مجله فرادرس توضیح دادیم از آيتم "Smooth Trajectory" در گزینه Graphical Rep تغییر دهید.
تعداد فریمها را میتوانید در کادر "Trajectory Step Size" تغییر دهید. حالت پیشفرض این گزینه ۱ است و به این معنی است که نرمافزار از تمام فریمها اسنپ شات میگیرد. اگر این عدد را به (۵) تغییر دهید، فریمهای مضرب ۵ و اگر به ۱۰ تغییر دهید، فریمهای مضرب ۱۰ در ساخت ویدیو استفاده میشود. با انتخاب آیتم "Delete Images" از گزینه "Movie Setting" در نورا بالایی این پنجره، پس از ساخت انیمیشن تصاویر اسنپ شات به صورت خودکار از سیستم شما پاک میشود.

از گزینه "Render" در نوار بالایی این پنجره روش ساخت انیمیشن را انتخاب کنید. استفاده از گزینه "Tachyon" با کیفیتترین خروجی انیمیشن را برای شما آماده میکند. قبل از شروع ساخت انیمیشن، از گزینه "Movie Setting" آيتم "Trajectory" را انتخاب کنید. با کلیک روی گزینه "Make Movie" پایین این پنجره ساخت انیمیشن شروع و با کلیک روی گزینه "Abort" ساخت انیمشین قبل از تکمیل شده تمام فریمها متوقف میشود. در پایان فرمت فیلم خروجی را با هر نرمافزاری که در اختیار دارید به mp4 تغییر دهید تا تصویر واضحی از مولکول در اختیار داشته باشید.
در این بخش از آموزش نرم افزار VMD نحوه ساخت انیمیشن با کیفیت از فایل تراجکتوری را توضیح میدهیم. برای ساختن انیمیشن با کیفیت بهتر میتوانید از نرمافزارهای جانبی ساخت فیلم مثل "Video Mach" استفاده کنید. در این روش گزینه "Delete Images" را قبل از ساخت انیمیشن در VMD انتخاب نکنید. در این حالت تصاویر با کیفیت بالا در سیستم شما ذخیره میشود. با آپلود کردن تصاویر در نرمافزار "Video Mach" میتوانید ویدیو شبیهسازی را با کیفیت بالا و فرمت دلخواه در اختیار داشته باشید.
جمعبندی آموزش نرم افزار VMD
در این مطلب از مجله فرادرس که با هدف آموزش نرم افزار VMD نوشته شده بود، توضیح دادیم که با استفاده از این نرمافزار میتوان فایلهای توپولوژی مولکول، تراجکتوری و فایلهای PDB پروتئینها را به شکل تصویری مشاهده کرد. پس از باز کردن نرمافزار VMD یک پنجره برای نمایش مولکول و یک پنچره برای کنترل نمایش مولکول و آنالیزها باز میشود.
با کلیک بر آیتم "New Molecule" در گزینه "File" پنجره کنترل، کاربر میتواند فایل مولکول را در نرمافزار لود کند. آیتمهای گزینه "Graphics" امکان تغییر ساختار و رنگ مولکولها را بری کاربر فراهم میکند و کلیک بر آیتم "Render" کاربر میتواند خروجی عکس مولکول را دریافت کند. یکی از مزیتهای این نرمافزار پشتیبانی زبانهای پایتون و Tcl است. در نتیجه کاربر میتواند از کدهای مورد نظر خود برای نمایش مولکول، تغییر رنگها یا انجام آنالیزها استفاده کند. به علاوه بخشی دستورات Tcl آنالیزهای کاربردی در بخش "Library" سایت رسمی نرمافزار به رایگان در اختیار کاربران قرار داده شده است.