تعیین گروه عاملی به کمک طیف سنجی مادون قرمز – از صفر تا صد
طیفسنجی، مطالعه برهمکنش تابشهای الکترومغناطیس با مولکولها است. تابشهای الکترومغناطیس شامل اشعه ایکس، ماروا بنفش، طیف مرئی، مادون قرمز، مایکروویو و امواج رادیویی میشود. تابش الکترومغناطیس را میتوان به صورت موجی با سرعت نور توصیف کرد. در نتیجه، این تابش، دارای طول موج و فرکانس خواهد بود. در این آموزش قصد داریم تا پس از یک مقدمه بسیار کوتاه از طیفسنجی، به تعیین گروه عاملی به کمک طیفسنجی مادون قرمز بپردازیم. از این روش طیف سنجی در شناسایی ترکیبات آلی استفاده فراوانی میشود.



مقدمهای بر طیفسنجی
در این بخش به طور خلاصه به طیفسنجی و روابط حاکم بر آن خواهیم پرداخت تا درک ادامه مطلب سادهتر باشد. همانطور که میدانید، انرژی تابشهای الکترومغناطیس به صورت کوانتیزه (کوانتیده) هستند.
این انرژی از رابطه زیر محاسبه میشود:
در رابطه بالا، ثابت پلانک و فرکانس است. با توجه به روابط فرکانس، رابطه بالا را میتوان به صورت زیر نوشت:
با تغییر نوشتار معادله قبل، به رابطهای بر حسب «عدد موج» (Wavenumber) میرسیم که نتیجه آن، رابطه زیر خواهد بود. این رابطه، انرژی تابش الکترومغناطیس را به عدد موج مرتبط میکند.
طیف الکترومغناطیس
طیف الکترومغناطیس شامل دامنه وسیعی از فرکانس و انرژی است. در تصویر زیر، طیف الکترومغناطیس به همراه طیف مرئی و طول موجهای مربوطه را مشاهده میکنید:

جذب تابش الکترومغناطیس
مولکولها میتوانند مقادیر گسسته و مشخصی از انرژی را دریافت کنند چراکه سطوح انرژی در مولکولها، به صورت کوانتیده است. برای تغییر انرژی یک مولکول، از به ، به اختلاف انرژی مشخصی نیاز داریم که فرکانس و طول موج مخصوص به خود را دارد. انرژی جذبشده توسط مولکول، موجب تغییر انرژی ارتعاشی یا الکترونی آن خواهد شد. به طور مثال، تابش فرابنفش، توزیع الکترونی در اوربیتالهای «پای» را تحت تاثیر قرار میدهد و تابش مادون قرمز (فروسرخ) موجب کشیدگی پیوندها و خمش در زاویه پیوندی میشود.
در انواع مختلفی از طیفسنجی، تابشی از منبع، از یک نمونه عبور میکند که ممکن است طول موجهای خاصی را جذب کند. با تغییر طول موج، آشکارساز، طول موج جذب شده از نمونه را مشخص میکند. در طول موج متناظر با اختلاف انرژی مورد نیاز مولکول برای تغییر، مولکول، تابش گسیل شده از منبع را جذب میکند. مقدار نور جذب شده توسط مولکول، به صورت تابعی از طول موج رسم میشود. در بیشتر طول موجها، مقدار تابش شناسایی شده توسط آشکارساز، مقدار برابری با مقدار گسیل شده از منبع دارد. در چنین شرایطی، مولکول، تابشی را جذب نکرده است. برای این طول موجها، نمودار، شامل یک خط افقی خواهد بود. که به آن، «خط پایه» (Base Line) میگویند.
زمانی که مولکول، طول موج خاصی را جذب کند، مقدار تابشی که به آشکارساز میرسد، کمتر از مقدار گسیلشده توسط منبع است و این اختلاف در نمودار به صورت یک پیک (قله) ثبت میشود. در طیفسنجی مادون قرمز، خط پایه در بالا و در روش «رزونانس مغناطیسی هسته» (NMR)، خط پایه در پایین قرار میگیرد.

طیفسنجی مادون قرمز و تعیین گروه عاملی
اتمهای یک مولکول نسبت به یکدیگر در جایگاه ثابتی قرار ندارند. مولکولها به طور ثابت، در فرکانسهای مختلفی ارتعاش میکنند که این ارتعاشات، وابسته به ساختار مولکول است. به طور مشابه، زاویه بین دو اتمی که به اتم مرکزی متصل شدهاند نیز بسته به ساختار مولکول، با فرکانسی کم و زیاد میشود. این فرکانسهای ارتعاش و خمش، با فرکانس نور در محدوده مادون قرمز متناظر هستند.
هر پیوندی در مولکول، با فرکانس خاصی، پرتو تابشی را جذب میکند. بنابراین، هر مولکول، طیف مادون قرمز مخصوص به خود را دارد. در تصویر زیر، طیف مادون قرمز ۱-متیل سیکلوپنتان را مشاهده میکنید. در محور افقی، عدد موج و واحد آن دیده میشود و محور عمودی نیز، درصد «عبور» (Transmittance) یا ترانسمیتانس را نشان میدهد. از آنجایی که عدد موج تابش جذب شده، به طور مستقیم به انرژی آن وابسته است، جذبهایی که عدد موج بالایی دارند (سمت چپ نمودار)، ارتعاشات مولکولی را نشان میدهند که به انرژی زیادی احتیاج دارند.
مجموعهای از دادههای جذب که در اعداد موج 1500 تا 2000 دیده میشوند، به محدوده «اثر انگشت» (Finger Print) طیف مادون قرمز موسوماند. در انتهای مبحث تعیین گروه عاملی، به این بخش بیشتر میپردازیم.
با توجه به اینکه هر مولکول، دارای «شاخصه» (Signature) مادون قرمز مخصوص به خود است، هر گروه عاملی نیز باندهای جذب مشخصی دارد که از طریق آن میتوان به تعیین گروه عاملی در یک مولکول پرداخت.

ارتباط بین فرکانسهای ارتعاشی و جرم اتمهای پیوندی
باوجود اینکه مجموعه ارتعاشات مولکولی، طیف مادون قرمز پیچیدهای را بدست میدهد، اما روشهای سادهای وجود دارند که جهت تفسیر و مقایسه جذبها به ما کمک میکنند. در یک تقریب اولیه، حرکت دو اتم پیوندی را میتوان به صورت مجزا در نظر گرفت. فرکانس ارتعاشی هر پیوند، به ثابت نیروی پیوند و جرم دو اتم، طبق رابطه زیر وابسته است:
ثابت نیروی پیوند تا حدودی به انرژی تفکیک پیوند نیز بستگی دارد. بنابراین، مقدار ثابت نیروی پیوند را میتوان با روند زیر بیان کرد:
پیوند سهگانه > پیوند دوگانه > پیوند یگانه
علاوه بر این، از آنجایی که پیوندهای قطبی مثل گروههای کربونیل، انرژی تفکیک پیوند بالاتری از پیوندهای دوگانه کربن-کربن دارند، ثابت نیرو برای گروه عاملی ، بزرگتر از گروه عاملی خواهد بود. به غیر از پیوندهای متصل به هیدروژن، اثر جرم اتمی، کمتر است. اثر جرم را میتوان با جایگذاری مقادیر آن در عبارت تعیین کرد. بنابراین خواهیم داشت:
بنابراین، «فرکانسهای کششی» (Stretching Frequencies) در پیوندهای ، و ، فاصله زیادی با فرکانسهای کششی در پیوندهای ، و دارند. کشش (کشیدگی) پیوندهای گروه اول، به انرژی بیشتری نیاز دارند و در نتیجه، در سمت چپ طیف مادون قرمز دیده میشوند.
در جدول زیر، محدوده جذب مادون قرمز برای برخی پیوندها نشان داده شده است که میتوان از آنها در تعیین گروه عاملی ترکیبات آلی بهره گرفت:
| نوع پیوند | محدوده جذب |
| 1300-800 | |
| 1900-1500 | |
| 2300-2000 | |
| 3650-2850 |
شناسایی و تعیین گروه عاملی هیدروکربنها
میدانیم که بسته به نوع پیوند در هیدروکربنها (یگانه، دوگانه و سهگانه)، آنها را به دو دسته هیدروکربنهای اشباع و غیراشباع تقسیمبندی میکنند. پیوندهای چندگانه را به کمک طیفسنجی مادون قرمز میتوان شناسایی کرد.
مولکولهای ۱-اوکتن و سیکلو اکتان، هردو فرمول مولکولی یکسانی به شکل دارند اما با نگاه به شکل مولکولی آنها میتوان فهمید که ۱-اوکتن، شامل پیوند دوگانهای است که در سیکلو اوکتان دیده نمیشود.

همانطور که وجود طیف جذب در طیفسنجی مادون قرمز مهم است، به طور معمول، عدم جذب نیز به همان میزان اهمیت دارد. در نتیجه، اگر یک گروه عاملی را به گروه دیگری تبدیل کنیم، مقایسه مادون قرمز واکنشدهنده و فرآورده به ما نشان میدهد که چقدر به هدف خود نزدیک شدهایم.
پیوند دوگانه کربن-کربن و پیوند کربن-هیدروژن به صورت هیبرید شده، ویژگیهای ساختاری هستند که در مولکول ۱-اوکتن وجود دارند اما در سیکلو اکتان دیده نمیشوند. از آنجایی که این ویژگیها سبب بوجود آمدن شاخصه جذب در یک گروه عاملی خواهند بود، در نتیجه، چنین مشخصهای را تنها در ۱-اوکتن شاهد هستیم. بنابراین، به سادگی میتوان با استفاده از طیفسنجی مادون قرمز به تعیین گروه عاملی و تفکیک این دو ترکیب پرداخت.
چنین ویژگی به طور مشابه در مقایسه بین دو ترکیب ۱-اوکتین و هیدروکربن ایزومری «بایسیکلو 0.3.3 اوکتان» قابل مشاهده است. ویژگیهای ساختاری در ۱-اوکتین عبارتند از: پیوند سهگانه کربن-کربن و پیوند کربن-هیدروژن هیبرید شده. انرژی مادون قرمز جذب شده پیوند کربن-هیدروژن، به هیبرید شدن اتم کربن بستگی دارد. قدرت پیوند کربن-هیدروژن را میتوان به صورت زیر توضیح داد:
در جدول زیر، فرکانسهای مربوط به گروههای عاملی آورده شده است که به کمک طیفسنجی مادون قرمز و این دادهها، میتوان به تعیین گروههای عاملی پرداخت:
| ترکیب آلی | گروه | عدد موج |
| آلکانها | 3000-2850 | |
| آلکنها | 3140-3080 | |
| 1670-1630 | ||
| آلکینها | 3320-3300 | |
| 2140-2100 | ||
| الکلها | 3600-3400 | |
| 1200-1050 | ||
| اترها | 1150-1070 | |
| آلدهیدها | 1725 | |
| کتونها | 1780-1700 |
پیوند هیبرید کربن-هیدروژن در هیدروکربنهای اشباع همچون اکتان، تابش مادون قرمز را در محدوده جذب میکند. همچنین، پیوند هیبرید کربن-هیدروژن در آلکنهایی مانند ۱-اوکتن، موجب جذب انرژی در عدد موج میشود. پیوند هیبرید کربن-هیدروژن نیز در مولکولی همچون ا-اوکتین، تابش مادون قرمز را در محدوده جذب میکند.
لازم به ذکر است که در هیدروکربن ایزومری بایسیکلی که در آن هیچ پیوند چندگانه کربن-کربن وجود نداشته باشد، اتمهای هیبرید شده و دیده نمیشوند و در نتیجه، محدوده جذب آن تنها در خواهد بود. در تصویر زیر طیف مادون قرمز ۱-اوکتین آورده شده است و به کمک آن میتوان مفاهیم گفته شده در خصوص تعیین گروه عاملی را به طور مجدد بررسی کرد.

هیدروکربنها را همچنین میتوان از طریق نحوه جذب پرتو مادون قرمز هم دستهبندی کرد. به این ترتیب، برای قدرت پیوند کربن-کربن میتوان روند زیر را در نظر گرفت:
پیوند سهگانه > پیوند دوگانه > پیوند یگانه
در نتیجه، عدد موج متناظر با کشش این پیوندها نیز با همین روند افزایش مییابد. هیدروکربنهای اشباع (سیرشده) اعم از آلکانها و سیکلوآلکانها، پیوندهای یگانه کربن-کربن زیادی دارند که پرتو را در محدوده جذب میکنند اما شدت این جذب بسیار پایین است.
پیوندهای یگانهای که در ترکیبات غیراشباع حضور دارند، مادون قرمز را در همین محدوده جذب میکنند. کشش و خمشهای مختلف دیگری نیز در این محدوده اتفاق میافتند که شدت آنها بیشتر است. بنابراین، در این محدوده، مقادیر محدودی جهت تعیین گروه عاملی وجود دارند. علاوه بر این، میدانیم که بیشتر ترکیبات آلی دارای پیوندهای یگانه هستند.
شناسایی و تعیین گروه عاملی ترکیبات شامل اکسیژن
بسیاری از گروههای عاملی که شامل اکسیژن هستند، جذب مادون قرمز مشخصی دارند که در جدول بالا آورده شدهاند. برای آلدهیدها و کتونها، محدوده جذب برابر با ذکر شده است. پیوند دوگانه کربن-اکسیژن در ترکیبات شامل گروه کربونیل، نسبت به پیوند یگانه کربن-اکسیژن، به انرژی بیشتری برای کشش پیوند نیاز دارد. این پیوندهای یگانه را میتوان در اترها و الکلها مشاهده کرد. بنابراین، آلدهیدها و کتونها، پرتو مادون قرمز را در اعداد موج بالاتری نسبت به الکلها و اترها جذب میکنند.
گروه کربونیل
میزان جذب در گروه عاملی کربونیل بسیار شدید است و به سادگی میتوان آنرا تشخیص داد زیرا در محدودهای از طیف مادون قرمز قرار میگیرد که «جذبهای تداخلی» (Conflicting Absorption) وجود ندارند. توجه داشته باشید که ارتعاش کششی پیوند دوگانه کربن-کربن، در عدد موج کمتری نسبت به ترکیبات کربونیل قرار دارد. در تصویر زیر، طیف معمول برای یک کتون (۲-هپتانون) نشان داده شده است که در آن، ارتعاش کششی کربونیل در اتفاق میافتد.

محل جذب گروه کربونیل، به اثرات القایی و رزونانس اتم متصل به کربن کربونیل وابسته است. گروه کربونیل، دارای دو ساختار رزونانس به شکل زیر است:

تعیین گروه عاملی الکلها و اترها
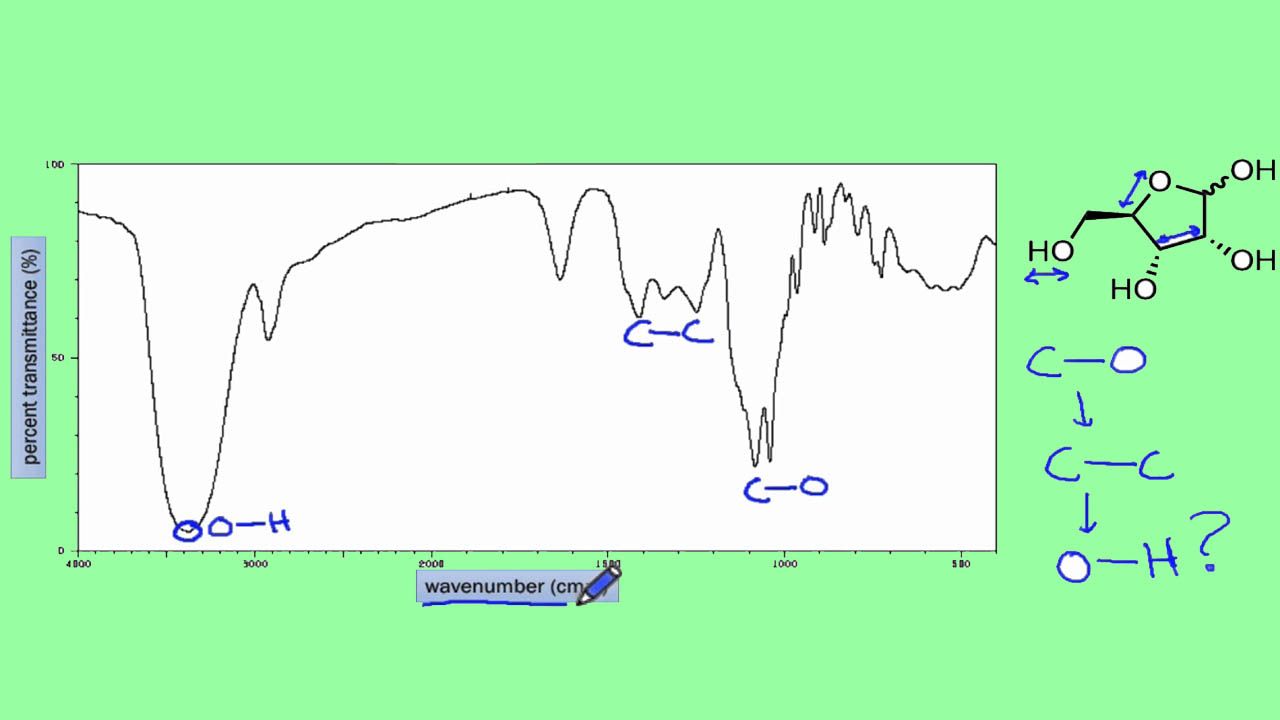
ارتعاش کششی پیوند کربن-اکسیژن در الکلها و اترها، در محدودهای ظهور میکند که با سایر جذبها تداخل دارد. البته، جذب پیوند یگانه کربن-اکسیژن، شدیدتر از جذب پیوند یگانه کربن-کربن است. حضور گروه هیدروکسیل را از طریق ارتعاش کششی پیوند اکسیژن-هیدروژن، بهتر میتوان تشخیص داد. این جذب به صورت یک پیک در محدوده دیده میشود که در تصویر زیر، برای ۱-بوتانول نشان داده شده است.
اترها را میتوان طی فرآیندی حذفی شناسایی کرد. اگر ترکیبی، اتم اکسیژن داشته باشد و طیف مادون قرمز آن، جذبی در محدوده مشخصه یک گروه کربونیل یا هیدروکسیل نداشته باشد، میتوان به این نتیجه رسید که ترکیب مورد نظر، یک اتر است.

تعیین گروه عاملی کربوکسیلیک اسیدها
کربوکسیلیک اسیدها، هم گروه هیدروکسیل و هم گروه کربونیل دارند. اگر مایعی خالص یا به صورت محلول داشته باشیم، جذب کشش پیوند در محدوده اتفاق میافتد. تحت این شرایط، یک کربوکسیلیک اسید به صورت دیمر با پیوند هیدروژنی حضور دارد. درنتیجه، محل جذب در کشش نزدیک به محدوده آلدهیدها و کتونها است. جذب کربوکسیلیک اسید، پهنای بسیار بیشتری از آلدهیدها و کتونها دارد و از این ویژگی میتوان برای تعیین گروه عاملی و شناسایی کربوکسیلیک اسیدها استفاده کرد.
علاوه بر این، جذب کشش پیوند در کربوکسیلیک اسیدها نیز از جمله ویژگیهای منحصر به فرد آن به شمار میآید که در همان محدوده طیف الکلها اتفاق میافتد. اما همانند جذب باید گفت که جذب پهنای زیادی را در محدوده در بر میگیرد. بنابراین، این جذب، به شدت با جذب در محدوده کشش پیوند همپوشانی دارد. در نتیجه، حضور یک طیف عریض در محدودههای و ، بوضوح بیانگر وجود یک کربوکسیلیک اسید است.

تعیین گروه عاملی استرها
استرهای ساده، جذبی در حدود دارند که عددی بالاتر از عدد موج در جذب آلدهیدها و کتونها است. استرها همچنین شامل جذب کشش پیوند کربن-اکسیژن در محدوده هستند. از آنجایی که در این محدوده، جذبهای دیگری هم دیده میشوند، از این دادهها تنها زمانی میتوان استفاده کرد که به کمک دادههایی دیگری همچون جذب کربونیل، به فرضِ حضور استر در ترکیب رسیده باشیم. به یاد داریم که الکلها، اترها و کربوکسیلیک اسیدها، همگی در این محدوده دارای جذب هستند. در تصویر زیر، طیف مادون قرمز اتیل استات نشان داده شده است.

تعیین گروه عاملی کربوکسیلیک اسید انیدرید
اسید آنیدرید در کربوکسیلیک اسیدها، دو ویژگی بارز دارد. ویژگی اول این است که دو جذب، یکی در حدود و دیگری در حدود خواهیم داشت که نتیجه حالات متقارن و نامتقارن ارتعاش هستند. دیگر اینکه انیدریدها به طور معمول، فرکانس قوی کشش (کشیدگی) پیوند در محدوده دارند. استیک انیدریدها نیز فرکانس قوی کشش پیوند دارند اما میزان جذب به مقدار منتقل شده است.

شناسایی و تعیین گروه عاملی ترکیبات شامل نیتروژن
پیشتر اشاره کردیم که فرکانسهای کشش پیوند با کربن-کربن، کربن-نیتروژن و کربن-اکسیژن فاصله زیادی دارد. کشش این پیوندها نیازمند انرژی بیشتری است و در سمت چپ طیف مادون قرمز دیده میشوند. خواهیم دید که این امر در خصوص آمینها و آمیدها نیز صدق میکند.
تعیین گروه عاملی آمینها
کشش پیوند ارتعاشی در آمینها، در محدوده پیوند اکسیژن-هیدروژن رخ میدهد. البته پیکهای مربوطه، با پیکهای طیف الکلها متفاوت است. میدانید که فرمول عمومی آمینهای نوع اول، دوم و سوم به ترتیب به صورت ، و نوشته میشود.
در آمینهای نوع اول، دو پیک در محدوده بین وجود دارد. همانند طیف اسید انیدریدها، پیک با بیشترین انرژی، نتیجه کشش متقارن و پیک با انرژی کمتر، نتیجه کشش نامتقارن است. مثالی از آنرا در تصویر زیر مشاهده میکنید که طیف ایزوپروپیل آمین را نشان میدهد. در این تصویر، کشش پیوند در محدوده اتفاق میافتد که نتیجه آن وجود پیکهای ضعیف است.

آمینهای نوع دوم، پیک تنها در محدوده دارند که از نمونههای آن میتوان به ان-متیل آنیلین اشاره کرد. در نتیجه، با مقایسه پیکها در طیفسنجی مادون قرمز به سادگی میتوان پیک در آمینهای نوع اول یا دوم را تشخیص داد.

تعیین گروه عاملی نیتریلها
باند جذب در نیتریلها با پیوند سهگانه نیتروژن-کربن، در محدوده دیده میشود که از آن برای تعیین گروه عاملی در نیتریلها استفاده میکنیم چراکه این منطقه، تداخلی با دیگر مناطق ندارد. در نتیجه، به راحتی میتوان به کمک طیفسنجی مادون قرمز آن را شناسایی کرد.

تغییر شکل حاصل از خمش
تمامی مواردی که تا اینجا در خصوص جذب مادون قرمز بیان کردیم، ارتعاشات کششی حاصل از حرکات اتمی در طول محور پیوند بودند. نوع دومی از ارتعاشات نیز وجود دارند که در جهت عمود بر پیوند اتفاق میافتند. به این نوع از ارتعاشات، «خمش» (Bending) میگویند که بیشتر در پیوندهای کربن-هیدروژن دیده میشوند. در خصوص آلکانها و ترکیبات آروماتیک، حرکات خمشی در بسیاری از پیوندهای کربن-هیدروژن، به صورت هماهنگ و یکی بعد از دیگری انجام میشوند که به کمک آنها میتوان اطلاعات مهمی در خصوص ساختارهای ایزومری پیدا کرد.
حالات خمشی مختلفی نیز برای گروههای آلکیلی وجود دارد. حرکات مختلف خمشی محتمل در ترکیبات آلی، بخش پیچیده در طیفسنجی مادون قرمز را تشکیل میدهد که پیشتر، نام «اثر انگشت» (Finger Print) را بر آن نهادیم.
آلکنها
آلکنها حالت (مد) خمش خارج از صفحه، در محدوده دارند. شدت این جذبها به اندازهای است که میتوان از آنها برای تعیین گروه عاملی استفاده کرد. آلکنهای ترمینال که پیوند دوگانه پای در آنها، انتهای زنجیر کربنی قرار دارد، الگوی جذب مشخص تولید میکنند. پیوندهای کربن-هیدروژن در آلکنهای ترمینال که به صورت وجود دارند، به طور هماهنگ دچار خمش میشوند و طیف جذبی در محدوده بوجود میآورند.
در ترکیباتی به شکل ، دو پیوند متیلن کربن-هیدروژن در محدوده جذب و پیوند دیگر در محدوده جذب میشوند. در نتیجه، هر دو نوع از آلکنهای ترمینال را میتوان شناسایی کرد.
آلکنهایی که به صورت ایزومرهای «سیس» (sis) و «ترانس» (trans) هستند، یک مد کربن-اکسیژن خارج از صفحه دارند. البته، جذب در ایزومرهای سیس، به طور معمول شناخته شده نیست.
| جذب | نوع پیوند |
| 995-985
910-905 |  |
| 895-885 |  |
| 980-965 |  |
| 690 |  |
| 840-790 |  |
حالات خمش در ترکیبات آروماتیک
جذب حاصل از خمش خارج از صفحه در ترکیبات آروماتیک، به الگوی جانشینی وابسته است. پیوندهای کربن-هیدروژن در اتمهای کربن مجاور، خمشی خارج از صفحه حلقه آروماتیک دارند. در جدول زیر میتوانید مقادیر جذب را به صورت تابعی از تعداد اتمهای هیدروژن مجاور ملاحظه کنید.
| تعداد اتمهای هیدروژن مجاور | عدد موج |
| 5 | 770-730 |
| 4 | 770-735 |
| 3 | 810-750 |
| 2 | 860-800 |
| 1 | 900-860 |
برای ترکیبات آروماتیک «تکاستخلافی» (Monosubstituted) همچون تولوئن، خمش خارج از صفحه پنج اتم هیدروژن مجاور، جذبی را ناحیه پدید میآورد.
برای مولکول ام-زایلین (۱و۳- دیمتیل بنزن)، خمش خارج از صفحه در روی میدهد. همچنین، برای اتمهای هیدروژن مجاور در ، و محدوده جذب برابر با خواهد بود و برای این مقدار برابر با است.

اگر این مطلب برای شما مفید بوده است، آموزشها و مطالب زیر نیز به شما پیشنهاد میشوند:
- مجموعه آموزشهای دروس شیمی
- مجموعه آموزشهای مهندسی شیمی
- آموزش شبیهسازی دینامیک مولکولی با نرمافزار GROMACS
- طیف سنجی مولکولی — به زبان ساده
- اصطلاحات کروماتوگرافی گازی — به زبان ساده
^^