شما در حال مطالعه نسخه آفلاین یکی از مطالب «مجله فرادرس» هستید. لطفاً توجه داشته باشید، ممکن است برخی از قابلیتهای تعاملی مطالب، مانند امکان پاسخ به پرسشهای چهار گزینهای و مشاهده جواب صحیح آنها، نمایش نتیجه آزمونها، پاسخ تشریحی سوالات، پخش فایلهای صوتی و تصویری و غیره، در این نسخه در دسترس نباشند. برای دسترسی به نسخه آنلاین مطلب، استفاده از کلیه امکانات آن و داشتن تجربه کاربری بهتر اینجا کلیک کنید.
بررسی تغییرات آنتالپی در ترمودینامیک | به زبان ساده
۱۲۴۵۷
۱۴۰۳/۰۹/۵
۱۴ دقیقه
PDF
آموزش متنی جامع
امکان دانلود نسخه PDF
در مباحث قبلی مجله فرادرس در خصوص اهمیت توابع حالت در انرژی درونی بحث کردیم. در این آموزش قصد داریم تا تغییرات آنتالپی را به عنوان یک تابع حالت در ترمودینامیک و در فرآیندهای همفشار و همدما بررسی کنیم. همچنین به طور کامل در خصوص ارتباط ظرفیتهای حرارتی Cp و Cv به بحث خواهیم پرداخت.
همانند انرژی درونی، آنتالپی را میتوان به صورت تابعی از دو متغیر از ۳ متغیر فشار، حجم و دما تعریف کرد. به هنگام بررسی توابع حالت در انرژی درونی دیدیم که بهتر است U را به صورت تابعی از دما و فشار بیان کنیم چراکه در نهایت به رابطه ΔU=qv رسیدیم. به طور مشابه، H را به صورت تابعی از دما و فشار بیان خواهیم کرد. حال این سوال پیش میآید که رابطه بین تغییرات آنتالپی با فشار و دما به چه صورت خواهد بود؟
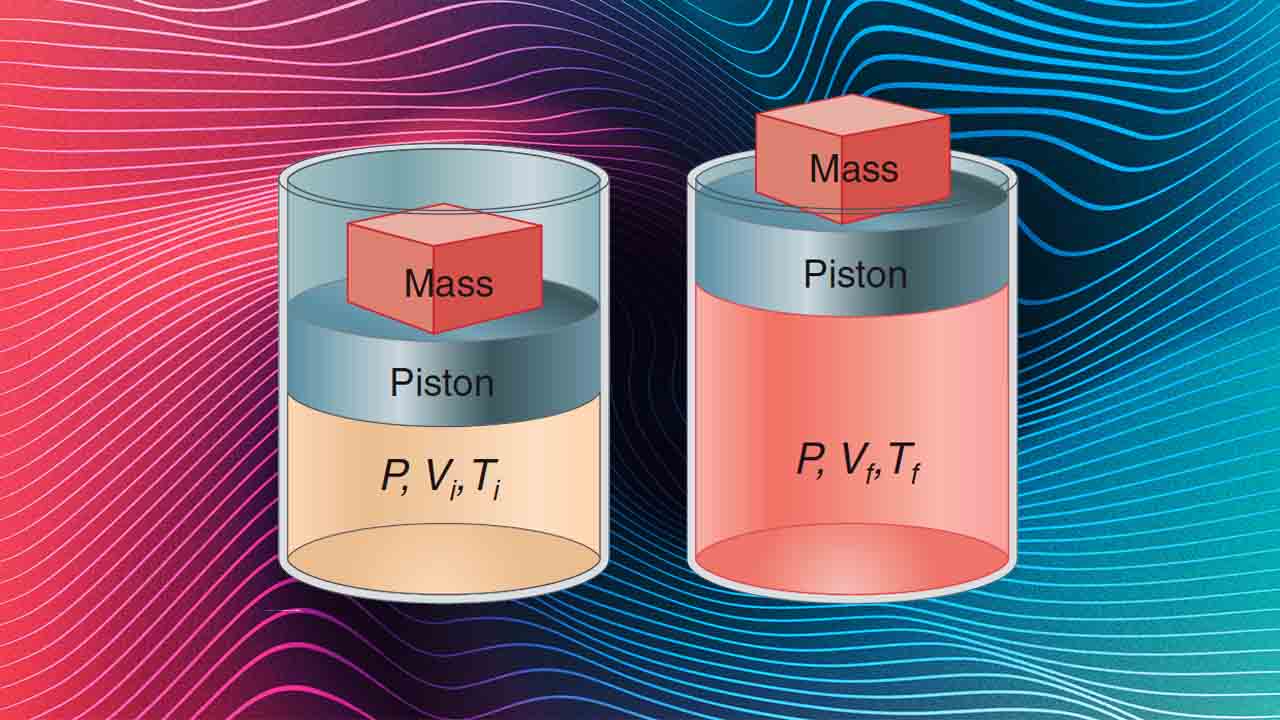
در ادامه این مطلب به تغییرات آنتالپی با دما در فشار ثابت خواهیم پرداخت. فرآیند همفشار که در تصویر زیر نشان داده شده است را در نظر بگیرید. برای این مساله، فشار را به صورت فشار خارجی (P=Pexternal) بیان کردهایم.
requirecanceldU=canceldqP−PdV
در این رابطه برای اینکه نشان دهیم q یک دیفرانسیل کامل نیست از خط استفاده کردهایم. با وجود اینکه انتگرالcanceldq وابسته به مسیر است اما تحت شرایط این مساله، مقدار منحصر به فردی دارد چراکه در اینجا، مسیر از قبل به صورت (P=Pexternal) تعریف شده است. با انتگرالگیری از دو طرف رابطه بالا، به دو رابطه زیر خواهیم رسید:
با توجه به اینکه P=Pf=Pi، رابطه بالا را میتوان به صورت یکی از ۲ شکل زیر نوشت:
(Uf+PfVf)−(Ui+PiVi)=qPΔH=qP
رابطه بالا نشان میدهد که مقدار تغییرات آنتالپی را در یک فرآیند همفشار داخل سیستم بسته و به همراه کار فشار-حجم، از طریق qp تعیین میشود. qp در اینجا، گرمای منتقل شده بین سیستم و محیط به صورت فرآیند فشار ثابت است. توجه داشته باشید که این رابطه با معادلات زیر که پیشتر در بحث اهمیت توابع حالت برای انرژی درونی مطرح کردیم، مشابه است:
intifdqV=intif(∂T∂U)VdTqV=ΔU
اما توجه داشته باشید که با توجه به شرایط ذکر شده، در یک فرآیند مشخص و در یک سیستم بسته، اگر تنها کار به صورت حجم-فشار باشد، ΔU=qV که در این حالت، فرآیند به صورت همحجم انجام میشود اما اگر فرآیند همفشار داشته باشیم، رابطه ΔH=qP برقرار خواهد بود. این دو رابطه، اساس روش کالریمتری (گرماسنجی) را تشکیل میدهند.
تغییرات آنتالپی و انرژی درونی در آزمایشگاه
از کاربردهای رابطه بدست آمده این است که میتوان از آن برای محاسبه آزمایشگاهی تغییرات آنتالپی و انرژی درونی ذوب و تبخیر استفاده کرد. ممکن است این سوال پیش بیاید که چطور ممکن است ذوب یا تبخیر در دمای ثابت رخ بدهند. «ذوب» (Fusion) و تبخیر، زمانی در دمای ثابت حاصل میشوند که سیستم را در یک فشار ثابت نگهداریم و گرما در طول مرز سیستم و محیط جریان پیدا کند.
در هر کدام از این تغییرات فاز، باید بر برهمکنشهای جاذبهای بین مولکولهای سیستم، غلبه کرد. در نتیجه، در هر یک از این حالتها مقدار q مثبت خواهد بود و CP به بینهایت میل میکند. با توجه به اینکه ΔH=qP، تغییرات آنتالپی ذوب و تبخیر را میتوان با اندازهگیری گرمای مورد نیاز برای این تغییر فاز در فشار ثابت، تعیین کرد. از آنجایی که رابطه ΔH=ΔU+Δ(PV) در فشار ثابت برقرار است، خواهیم داشت:
تغییرات حجم در تبخیر را به صورت ΔVvaporization=Vgas−Vliquid بیان میکنیم که مقدار آن مثبت است. در نتیجه، تغییرات انرژی درونی تبخیر، کمتر از تغییرات آنتالپی تبخیر خواهد بود یعنی ΔUvaporization<ΔHvaporization. رابطهای مشابه رابطه بالا را نیز میتوان برای تغییرات آنتالپی و انرژی درونی ذوب نوشت. توجه داشته باشید که تغییر حجم ناشی از ذوب، بسیار کمتر از تغییر حجم ناشی از تبخیر است و میتواند مقداری مثبت یا منفی اختیار کند. به همین دلیل، رابطه زیر را برای ذوب در نظر میگیریم:
ΔUfusion≈ΔHfusion
از آنجایی که H، نوعی تابع حالت به شمار میآید، dH نیز دیفرانسیل کامل خواهد بود و از این طریق میتوانیم (∂T∂H)P را به یک مقدار قابل اندازهگیری مرتبط کنیم. dH را در نهایت به صورت رابطه زیر خواهیم نوشت:
dH=(∂T∂H)PdT+(∂P∂H)TdP
با توجه به اینکه فشار ثابت داریم، dP برابر با صفر خواهد بود همچنین دیدیم که dH=canceldq، در نتیجه رابطه بالا را به شکل زیر خواهیم نوشت:
dqP=(∂T∂H)PdT
این رابطه در نهایت سبب ارائه معادلهای برای ظرفیت گرمایی در فشار ثابت (CP) خواهد شد که به صورت زیر تعریف میشود:
CP=dTcanceldqP=(∂T∂H)P
رابطه بالا نیز همچون CV در بحث انرژی درونی، ناملموس به نظر میآید اما کمیتی است که به راحتی میتوان آنرا اندازهگیری کرد. برای اندازهگیری آن باید گرمای جریان یافته به محیط در فرآیند فشار ثابت به همراه تغییر دمای حاصل شده را اندازهگیری کنیم. این اندازهگیری باید به صورت مقادیر دیفرانسیلی و بسیار کوچک درنظر گرفته شود تا نسبت حدی زیر برقرار باشد:
dT→0lim(canceldq/dT)P
همانند CP، CV نیز نوعی خاصیت مقداری به شمار میآید که در هر ماده متفاوت است. بمنظور محاسبه تغییرات آنتالپی با دما، وابستگی دمایی CP را باید بشناسیم. برای یک فرآیند همفشار که در آن، هیچ تغییر فاز و واکنش شیمیایی صورت نگیرد، تغییرات آنتالپی به صورت زیر تعریف میشود:
ΔHP=∫TiTfCP(T)dT=n∫TiTfCPm(T)dT
اگر بازه دمایی به اندازه کافی کوچک باشد، میتوان CP را عددی ثابت فرض کرد، در چنین شرایطی، رابطه بالا به رابطه زیر تبدیل میشود:
ΔHP=CPΔT=nCPmΔT
مثال برای محاسبه تغییرات آنتالپی در فشار ثابت
143 گرم گرافیت را از دمای 300 تا 600 کلوین در فشار ثابت حرارت میدهیم. در این بازه دمایی، CPm به کمک رابطه زیر تعیین میشود:
ΔH و qP را محاسبه کنید. اگر وابستگی دمایی CPm را در نظر نگیریم و فرض کنیم که این عدد برای دمای 300 کلوین، ثابت میماند، میزان خطای اندازهگیری تغییرات آنتالپی چه میزان خواهد بود؟
اگر فرض کرده بودیم که CPm در دمای 300 کلوین ثابت میماند، مقدار محاسبه شده آن برابر با 8.617Jmol−1K−1 میبود که در اینصورت، تغییرات آنتالپی به صورت زیر محاسبه میشد:
$$\begin{equation} \require {cancel} d q = C _ {V} d T + \left ( \frac { \partial U } {\partial V } \right ) _ { T } d V + P _ { \text {external} } d V \end {equation}$$
فرآیندی با فشار ثابت و P=Pexternal را در نظر بگیرید. در این شرایط، رابطه بالا به شکل زیر تبدیل خواهد شد:
dqP=CVdT+(∂V∂U)TdV+PdV
با توجه به اینکه dqp=CpdT، با تقسیم دو طرف رابطه بر dT و تبدیل نسبت dV/dT به مشتق جزئی در فشار ثابت، خواهیم داشت:
رابطه بالا نمونهای از کاربرد ترمودینامیک در مرتبط کردن مشتقات جزئی با دادههای آزمایشگاهیِ در دسترس است. تفاوت بین CPm و CVm در دمایی مشخص را با دانستن حجم مولی، ضریب انبساط حجمی همفشار و تراکمپذیری همدما میتوان تعیین کرد. رابطه بالا در گازهای حقیقی و ایدهآل و همچنین در مایعات و جامدات، در غیاب تغییرات فاز و واکنشهای شیمیایی بکار گرفته میشوند. از آنجایی که برای گازهای واقعی و ایدهآل، عبارات κ و β همواره مثبت هستند، Cp−CV نیز برای این مواد مقدار مثبتی دارد. در ابتدا Cp−CV را برای یک گاز ایدهآل محاسبه میکنیم و سپس این کار برای مایعات و جامدات انجام خواهد شد. برای یک گاز ایدهآل، (∂V∂U)T=0، همچنین عبارت زیر نیز برای آن برقرار است:
T(∂T∂P)V(∂T∂V)P=T(VnR)(PnR)=nR
در نتیجه به رابطه زیر خواهیم رسید:
CP−Cv=nR
مشتق جزئی (∂T∂V)P=Vβ برای مایعات و جامدات، نسبت به گازها عبارت بسیار کوچکی است، بنابراین به طور کلی داریم:
CV≫[(∂V∂U)T+P](∂T∂V)P
بنابراین برای مایعات و جامدات، مقدار Cp تقریبا با CV مساوی است.
تغییرات آنتالپی با فشار در دمای ثابت
در بخش قبل یاد گرفتیم که چطور آنتالپی با دما و در فشار ثابت تغییر میکند. برای محاسبه نحوه تغییرات آنتالپی به هنگام تغییر فشار و دما، باید (∂P∂H)T را محاسبه کنیم. این مشتق جزئی را نمیتوان مثل (∂T∂H)P به راحتی به صورت مستقیم محاسبه کرد. همانطور که خواهیم دید، برای بسیاری از فرآیندها با تغییر فشار و دما، (∂H/∂T)PdT≫(∂H/∂P)TdP خواهد بود و میتوان از وابستگی فشاری H نسبت به وابستگی دمایی، صرف نظر کرد.
با این وجود، دانستن اینکه (∂P∂H)T مقداری غیر صفر دارد برای فهم طرز کار یخچالها و مایع کردن گازها ضروری است. بحثی که در ادامه پیش خواهیم برد را میتوان در گازها، مایعات و جامدات بکار برد. با داشتن رابطه H=U+PV، کار خود را با نوشتن dH به شکل زیر آغاز میکنیم:
dH=dU+PdV+VdP
با جایگذاری مقادیر دیفرانسیلی dU و dH به رابطه زیر میرسیم:
در یک فرآیند همدما، dT=0 و رابطه بالا به شکل زیر تغییر خواهد کرد. با استفاده از رابطهای که در بحث تغییرات انرژی درونی برای (∂V∂U)T بیان کردیم، خواهیم داشت:
با بکارگیری قانون ضرب سهگانه، فرمولبندی دیگری را نیز میتوان برای رابطه بالا ارائه داد. این رابطه به شرطی که تغییرات فاز و همچنین واکنش شیمیایی نداشته باشیم، در تمامی سیستمها اعم از مواد خالص و مخلوط (با ترکیب ثابت) صدق میکند.
این نتیجه را میتوانستیم به طور مستقیم از تعریف H=U+PV نیز بدست آوریم. برای یک گاز ایدهآل، انرژی درونی، تنها تابعی از دما است و بنابراین PV=nRT و در نتیجه، آنتالپی نیز تابعی از دما خواهد بود و خواهیم داشت:
(∂P∂H)T=0
تغییرات آنتالپی در مایعات و جامدات
از آنجایی که در مثال قبل دیدیم برای یک گاز ایدهآل، H تنها تابعی از دما است، بنابراین در محاسبه تغییرات آنتالپی خواهیم داشت:
با توجه به اینکه در یک گاز ایدهآل، آنتالپی تابعی از دما است، رابطه فوق حتی زمانی که فشار ثابت نباشد نیز در یک گاز ایدهآل صدق میکند. این نتیجه را میتوان از مفهوم تابع پتانسیل نیز دریافت کرد. به دلیل اینکه مولکولها در یک گاز ایدهآل یکدیگر را جذب یا دفع نمیکنند، هیچ انرژی برای تغییر فاصله متوسط جدایش (افزایش یا کاهش فشار) نیاز نیست.
حال، رابطهای که برای محاسبه تغییرات آنتالپی در دمای ثابت بدست آوردیم را برای سایر سیستمها بکار میبریم. دیدیم که برای یک گاز ایدهآل، تغییرات آنتالپی نسبت به فشار در دمای ثابت، برابر با صفر بود. در جامدات و مایعات، در دماهای کمتر از 1000 کلوین، حاصلضرب Tbeta بسیار کمتر از مقدار ۱ خواهد بود و این گفته، در جداول ترمودینامیکی قابل مشاهده است. بنابراین، برای جامدات و مایعات، با تخمین مناسبی، رابطه (∂P∂H)T≈V برقرار است و dH را برای سیستمهایی که تنها شامل مایع یا جامد باشند، میتوان به شکل زیر نوشت:
dH≈CpdT+VdP
مثال برای محاسبه تغییرات آنتالپی در مایعات
اگر ۱۲۴ گرم متانول مایع در فشار 1bar و دمای ۲۹۸ کلوین به فشار 2.55ˉ و دمای ۴۲۵ کلوین رسد، تغییرات آنتالپی را برای این ماده محاسبه کنید. تحت این شرایط:
با توجه به اینکه H نوعی تابع حالت به شمار میآید، هر مسیر انتخاب شده بین حالات اولیه و نهایی، به یک ΔH ختم میشود. به همین دلیل، برای محاسبه، مسیر زیر را انتخاب میکنیم:
توجه داشته باشید که در این سوال، سهم تغییرات دما در تغییرات آنتالپی بسیار بیشتر از سهم فشار است.
مثال بالا نشان میدهد که به دلیل اینکه حجمهای مولی کم جامدات و مایعات، نسبت به تغییرات فشار، تغییرات آنتالپی با شدت بیشتری به هنگام تغییر دما رخ میدهند. در بیشتر موارد، برای جامدات و مایعات، آنتالپی را میتوان تنها تابعی از دما دانست. مواردی که از این حالت پیروی نمیکنند شامل کاربردهای ژئوفیزیکی و نجوم است که تغییرات شدید فشار را تجربه میکنند.
نتیجه کلی را میتوان اینگونه بیان کرد: در بیشتر شرایطی که یک شیمیدان در آزمایشگاه تجربه میکند، آنتالپی را میتوان برای مایعات و جامدات، تنها تابعی از دما دانست. تخمین مناسبی از تغییرات آنتالپی به صورت رابطه زیر خواهد بود.
وابستگی آنتالپی به فشار را برای گازهای ایدهآل، در مبحث آزمایش ژول تامسون مورد بررسی قرار میگیرد. توجه داشته باشید که رابطه بالا زمانی کاربرد دارد که هیچ واکنش شیمیایی و هیچ تغییر فازی نداشته باشیم.
«سهیل بحرکاظمی» دانشآموخته کارشناسی ارشد رشته مهندسی نفت از دانشگاه علوم و تحقیقات تهران است. به عکاسی و شیمی آلی علاقه دارد و تا امروز تولید مطالب متنوعی از مجله فرادرس را در حوزههای شیمی، هنر و بازاریابی به عهده داشته است. او اکنون به عنوان مدیر مجله علمی-آموزشی فرادرس فعالیت میکند.
شما در حال مطالعه نسخه آفلاین یکی از مطالب «مجله فرادرس» هستید. لطفاً توجه داشته باشید، ممکن است برخی از قابلیتهای تعاملی مطالب، مانند امکان پاسخ به پرسشهای چهار گزینهای و مشاهده جواب صحیح آنها، نمایش نتیجه آزمونها، پاسخ تشریحی سوالات، پخش فایلهای صوتی و تصویری و غیره، در این نسخه در دسترس نباشند. برای دسترسی به نسخه آنلاین مطلب، استفاده از کلیه امکانات آن و داشتن تجربه کاربری بهتر اینجا کلیک کنید.




















بسیار خوب ما هر وقت از این سایت استفاده میکنیم