



داکینگ مولکولی چیست؟ – کاربردها، روش ها و نرم افزارها – به زبان ساده
داکینگ مولکولی یک ابزار کلیدی در زیست شناسی مولکولی ساختاری و طراحی داروهای رایانهای است. هدف اتصال لیگاند - پروتئین این است که حالت یا حالتهای متصل کننده اصلی لیگاند را با پروتئینی دارای ساختار سه بعدی شناخته شده پیش بینی کنیم. در این مطلب به تعریف داکینگ مولکولی و انواع آن و همچنین روشهای انجام آن با استفاده از نرم افزارهای مختلف میپردازیم، علاوه بر این منابع یادگیری داکینگ مولکولی را نیز معرفی میکنیم.

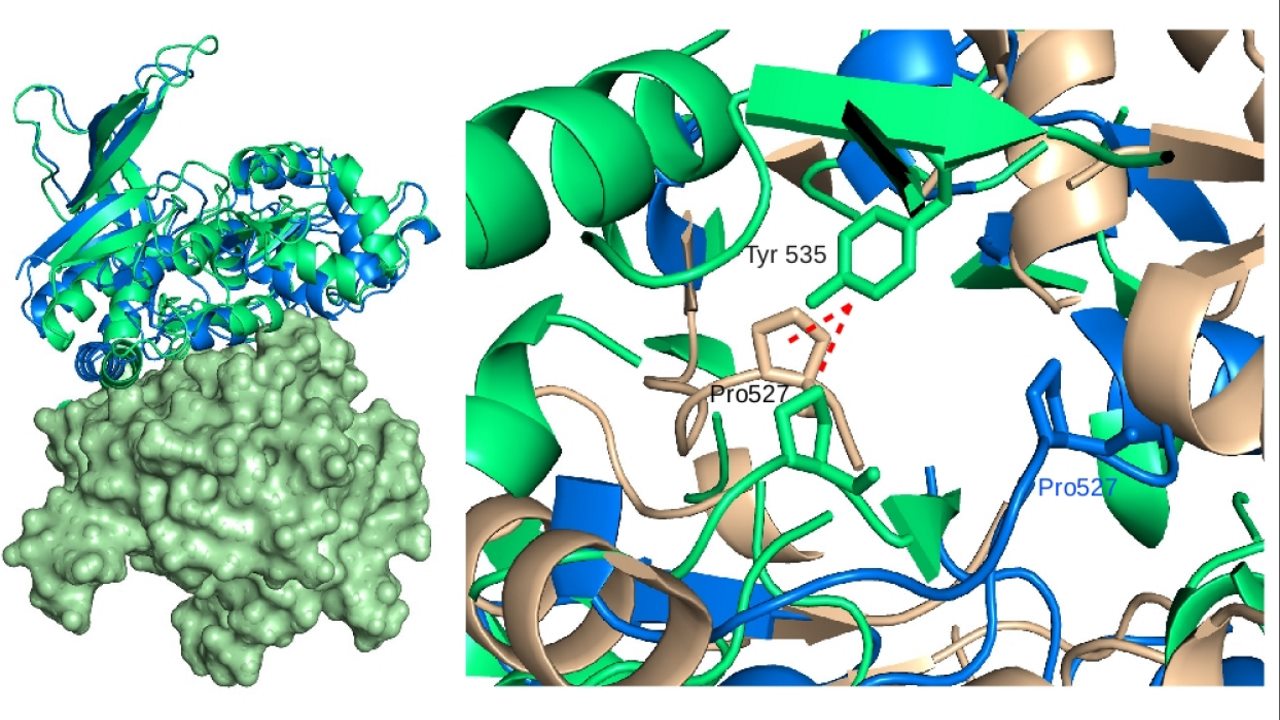


داکینگ مولکولی چیست؟
«داکینگ مولکولی» (Molecular Docking) یا متصل کردن مولکولی یک روش محاسباتی است که برای پیشبینی برهمکنش دو مولکول متصل شونده استفاده میشود. داکینگ مولکولی به دلیل توانایی آن در پیشبینی اتصال و ترکیب لیگاندهای مولکولی کوچک به محل اتصال مناسب، یکی از متداولترین روشها در طراحی دارو است. تعیین مشخصات و رفتار اتصال دهنده نقش مهمی در طراحی منطقی داروها و همچنین روشن ساختن فرایندهای بیوشیمیایی اساسی دارد.
داکینگ یک روش محاسباتی برای جستجوی یک لیگاند مناسب است که هم از نظر انرژی و هم از نظر هندسی با محل اتصال پروتئین مطابقت دارد. به عبارت دیگر، در مطالعات داکینگ این مسئله مطرح است که چگونه دو یا چند مولکول در اتصال با هم متناسب هستند و مشابه حل کردن یک پازل سه بعدی است.

فرایند داکینگ مولکولی چگونه است؟
داکینگ مولکولی به عنوان ابزار بسیار کارآمد برای کشف داروهای جدید برای هدف قرار دادن پروتئین ثابت شده است. در بین انواع مختلف داکینگ، اتصال پروتئین - لیگاند به دلیل کاربرد آن در صنعت پزشکی از اهمیت خاصی برخوردار است. اتصال پروتئین - لیگاند به جستجوی سازگاری دقیق لیگاند در یک پروتئین هدف هنگامی که ساختار پروتئینها مشخص است اشاره دارد. میتوان داکینگ مولکولی را به عنوان یک مسئله قفل و کلید در نظر گرفت که در آن میخواهیم جهت نسبی صحیح کلید را پیدا کنیم که قفل را باز میکند (جایی که در سطح قفل سوراخ کلید وجود دارد، پس از قرار دادن کلید به کدام جهت بپیچد و غیره)، که میتوان پروتئین را قفل و لیگاند را کلید در نظر گرفت.
در حال حاضر، مکانیک مولکولی پایه اکثر برنامههای داکینگ است. مکانیک مولکولی شامل توصیف یک سیستم چند اتمی با استفاده از فیزیک کلاسیک است. پارامترهای تجربی مانند بارها، زاویههای پیچشی و هندسی برای محدود کردن تفاوت بین دادههای تجربی و پیش بینیهای مکانیک مولکولی استفاده میشود. به دلیل کمبودها و محدودیتهای پارامترهای تجربی، معادلات ریاضی اغلب ممکن است بر اساس محاسبات نظری مکانیک کوانتوم نیمه تجربی و بدوی پارامتر شوند. به این ترتیب، میدانهای نیروی مولکولی مجموعهای از معادلات با پارامترهای مختلف با هدف نهایی توصیف سیستمها هستند. با استفاده از میدانهای نیرو، مدل مولکولی و پروتئینی در اوایل دهه 1980 انجام شد.
هدف از گسترش طبیعی این روشها مدل سازی فرآیندهای مولکولی مانند اتصال پروتئین به لیگاند بود. دو روش کلی توسعه داده شد که اولین رویکرد با در نظر گرفتن سفت و سخت بودن جسم است، در این مدل، لیگاند و گیرنده به عنوان دو جسم مستقل در نظر گرفته میشوند که بر اساس شکل و حجم یکدیگر را تشخیص میدهند. روش دوم، داکینگ انعطاف پذیر است که این رویکرد یک اثر متقابل تشخیص پروتئین - لیگاند بر ترکیب هر قسمت را در نظر میگیرد. طی 15 سال گذشته، تعداد مقالات مرتبط با اتصال مولکولی به میزان قابل توجهی افزایش یافته است. تحقیقات داکینگ مولکولی بر شبیه سازی محاسباتی فرآیند تشخیص مولکولی متمرکز است. هدف آن دستیابی به یک ترکیب بهینه برای پروتئین و لیگاند و یافتن جهت نسبی بین پروتئین و لیگاند است تا انرژی آزاد سیستم کلی به حداقل برسد.

سخت افزار مورد نیاز داکینگ مولکولی
قبل از پرداختن به جزئیات علمی روش داکینگ، در مورد الزامات سخت افزاری عمومی برای اجرای کارآمد داکینگ توضیح خواهیم داد. معمولاً محاسبه داکینگ با توجه به CPU سیستم شما زیاد در نظر گرفته نمیشود زیرا لیگاندها ممکن است در عرض چند دقیقه متصل شده و ارزیابی شوند. در حال حاضر، تقریباً هر رایانه شخصی (یا لپ تاپ) به اندازه کافی شایسته است که یک رشته عملیات کوچک داکینگ (حدود 500 - 1000 ترکیب) را در یک زمان معقول اجرا کند.
با این حال، غربالگری مجازی مخازن مبتنی بر داکینگ با استفاده از پایگاههای داده عمومی میتواند به سرعت افزایش یابد (بیش از ۱ میلیون ترکیب)، که نیاز به منابع محاسباتی بیشتری دارد تا طی چند هفته به پایان برسد. در جدول زیر دستورالعملهای کلی توصیه شده برای تهیه سخت افزار لازم قبل از اجرای داکینگ مولکولی ارائه شده است.
| مقادیر توصیه شده | حداقل قابل پذیرش | مشخصات |
| x86_64 | i686 | Processor architecture |
| ۳/۴ گیگاهرتز یا بالاتر | ۱ گیگا هرتز | Processor clock rate |
| 8 | 2 | Number of threads |
| 4 گیگابایت یا بالاتر | ۱ گیگا بایت | RAM |
| 10 گیگابایت یا بالاتر | ۱ گیگا بایت | HDD |
نرم افزارهای داکینگ مولکولی
چندین سرور، مجموعه و نرم افزار برای اتصال مولکولی وجود دارد. هر ابزار از میدانهای نیرو و الگوریتمهای متفاوتی برای ایجاد ژست، غربال گری و محاسبه فعل و انفعالات گیرنده - لیگاند استفاده میکند. علیرغم این واقعیت که تعداد زیادی برنامه قوی برای داکینگ وجود دارد، پژوهشگر باید در نظر داشته باشد که همه الگوریتمهای اتصال برای هر سیستم مناسب نیست و به طور کلی توصیه میشود از بیش از یک برنامه اتصال استفاده کنید: مطالعات مختلف نشان داده است که به طور کلی، اجتماعی از پروتکلهای مختلف داکینگ، ارزیابی بهتری از فعل و انفعالات لیگاند - پروتئین و رتبه بندی ژست قابل اطمینان تری را به همراه دارد. جدول زیر لیستی کوتاه از برنامههای اصلی داکینگ مولکولی، شامل الگوریتمها و اطلاعات کلی را ارائه میدهد.
| اسامی نرم افزارها | الگوریتم جستجو | نوع نرم افزار |
| AUTODOCK4 | الگوریتم ژنتیک «لامارکیان» (Lamarckian) | آکادمیک |
| DOCK | «تطبیق شکل» (Shape matching) | آکادمیک |
| OEDOCKING | «تطبیق شکل» (Shape matching) | اکادمیک |
| FLEKSY | «مبتنی بر گروه» (Ensemble-based) | تجاری |
| SWISSDOCK | بهینه سازی تکاملی | آکادمیک |
| GOLD | الگوریتم ژنتیک | تجاری |
| VINA | «بهینه سازی محلی» (Local optimization) | آکادمیک |
| RDOCK | ترکیبی | آکادمیک |
| LEDOCK | انلینگ شبیه سازی شده | آکادمیک |
| HADDOCK | ترکیبی | آکادمیک |
| MOE | ترکیبی | تجاری |
| SURFLEX-DOCK | «تطبیق شکل» (Shape matching) | تجاری |
| FLEXX | «تطبیق شکل» (Shape matching) | تجاری |
| FITTED | ترکیبی | تجاری |
| LIGANDFIT | «تطبیق شکل» (Shape matching) | تجاری |
بهترین نرم افزار برای داکینگ مولکولی کدام است؟
نرم افزارهای آکادمیک یا رایگان به دلیل سهولت دسترسی، نقطه شروع خوبی است. متأسفانه اکثر این برنامهها دارای یک منحنی یادگیری کند هستند (به عنوان مثال DOCK ، rDock و PLANTS). یکی از این دلایل عدم تسلط کاربران عادی به استفاده از سیستم عاملهای لینوکس و شبه اونیکس است. این بدان معنا نیست که نرم افزار اتصال به سیستم عاملهای مبتنی بر UNIX منحصر به فرد است، فقط برخی برنامهها برای اجرای صحیح آنها به مترجمان مانند Cygwin یا Git وابسطه هستند. با این وجود، نصب و استفاده از چنین ابزارهایی ممکن است به ویژه برای کاربران مبتدی مشکل ساز باشد. به عبارت دیگر نرم افزارهای داکینگ مولکولی برای ویندوز طراحی نشده اند و بسیاری از مشکلات افراد مبتدی در صورتی که از ابتدا با لینوکس کار کنند به وجود نخواهد آمد.
با این وجود، اگر افراد نیازمند نرم افزارهای هماهنگ با ویندوز برای انجام داکینگ مولکولی هستند ابزارهایی مانند، Autodock و Vina (به همراه AutodockTools) برای این پلتفرم در دسترس هستند. گزینه دیگر برای ویندوز LeDock است که دارای GUI ساده و در عین حال موثر است (اگرچه غربالگری مجازی برای لینوکس اجرا شده است). در صورت استفاده از بیش از یک برنامه، میزان موفقیت بالاتری در فرایند داکینگ مولکولی وجود دارد. با این حال، همانطور که در ادامه مورد بحث قرار گرفته است انتخاب مجموعهای از برنامههای مختلف باید با دقت انجام شود تا از پاسخ مثبت کاذب جلوگیری شود. توصیههای زیر در مورد استفاده از برخی از برنامههای داکینگ میتواند مفید باشد.
- Vina یک نقطه شروع عالی است و ممکن است به دلیل عملکرد خوب در برابر چندین خانواده پروتئینی به عنوان نرم افزار بسیار کاربردی شناخته شود. این برنامه با توجه به مشتقات مختلفی که دارد ویژگیهای اضافی و سهولت استفاده را به کاربر ارائه میدهد.
- LeDock یک برنامه نسبتا جدید است. استفاده ساده و محاسبات سریع از نقاط قوت اصلی آن است. همچنین، ژستها خوشه بندی قابل توجهی را نشان میدهند. این برنامه معمولا همراه LePro بوده که یک ماژول آماده سازی است.
- MOE رابط کاربری گرافیکی خوبی دارد و بسیار بصری است. همچنین برای داکینگ، چندین الگوریتم جستجو و توابع نمره گذاری دارد. علاوه بر این، MOE از ابزارهای اضافی مانندMOPAC ، GAMESS ، Gaussian ، NAMD ، FlexX ، GOLD و OMEGA پشتیبانی میکند. از نقاط ضعف آن نتایج غیرمنتظره با ماژول آماده سازی پروتئین بوده زیرا در برخی موارد هزینههای اشتباه را به همراه دارد.
- DOCK یکی از اولین برنامههای داکینگ مولکولی بوده که توسعه داده شده است. نسخه فعلی آن DOCK6، توابع امتیاز دهی جدید و روشهای تجزیه و تحلیل را معرفی میکند. همانطور که قبلاً ذکر شد، این یکی از معدود برنامههایی با پیاده سازی GPU برای نمره گذاری AMBER و محاسبه PBSA/GBSA برای مجتمعهای لیگاند - پروتئین است. از آنجا که برای کار با DockPrep طراحی شده است، دانلود و استفاده از USCF Chimera مورد نیاز است، با این وجود این امر امکان معرفی یک ابزار بسیار مفید و کارآمد دیگر را میدهد. معایب نسبی DOCK این است که فقط در لینوکس اجرا میشود، در مقایسه با سایر برنامهها نیاز به تجربهای در تدوین نرم افزار و عملکرد ضعیف نسبی امتیازبندی پیش فرض دارد.
- rDock یک برنامه قوی مبتنی بر لینوکس با عملکرد قابل توجه است. منحنی یادگیری کند آن ممکن است یکی از ضعفهای اصلی آن باشد به ویژه اگر کاربر نهایی هرگز از لینوکس استفاده نکرده باشد، ممکن است مشکلاتی را ایجاد کند. علاوه بر این، rDock ماژول آماده سازی ندارد.
مکانیک داکینگ مولکولی
برای اجرای صفحه داکینگ، اولین عامل مورد نیاز ساختار پروتئین مورد نظر است. معمولاً ساختار با استفاده از تکنیک بیوفیزیکی مانند کریستالوگرافی اشعه ایکس، طیف سنجی NMR یا میکروسکوپ الکترونی کرایو (Cryo - EM) تعیین شده است، اما میتواند از ساختار مدل سازی همولوژی نیز ناشی شود. این ساختار پروتئینی و یک پایگاه داده از لیگاندهای بالقوه به عنوان ورودی یک برنامه داکینگ است. موفقیت یک برنامه داکینگ به دو جزء بستگی دارد: الگوریتم جستجو و عملکرد امتیازدهی. در ادامه به بررسی هر کدام میپردازیم.
الگوریتم جستجو
فضای جستجو از نظر تئوری شامل تمام جهت گیریها و تطابق پروتئین همراه با لیگاند است. با این حال، در عمل با منابع محاسباتی فعلی، امکان جستجوی جامع فضای جستجو وجود ندارد. این جستجو شامل شمارش همه حالتهای احتمالی هر مولکول (مولکولها پویا هستند و در مجموعهای از حالتهای تطبیقی وجود دارند) و همه جهتهای چرخشی و برگشتی لیگاند نسبت به پروتئین است. بیشتر نرم افزارهای داکینگ در حال استفاده کل فضای سازنده لیگاند (لیگاند انعطاف پذیر) را در بر میگیرد و چندین برنامه سعی میکنند یک گیرنده پروتئینی انعطاف پذیر را مدل سازی کنند. به هر عکس فوری این جفت (لیگاند و پروتئین) به عنوان «ژست» (pose) قرارگیری مولکولها اشاره میشود. انواع استراتژیهای جستجوی سازگاری برای لیگاند و گیرنده اعمال شده است که شامل موارد زیر هستند:
- جستجوهای پیچشی سیستماتیک یا تصادفی در مورد پیوندهای قابل چرخش.
- شبیه سازی دینامیک مولکولی
- الگوریتمهای ژنتیک برای تکامل سازگاریهای جدید با انرژی کم و در جایی که نمره هر ژست به عنوان عملکرد مناسب برای انتخاب جهت تکرار بعدی عمل میکند.
الگوریتمهای جستجو بر اساس عوامل مختلفی از جمله انعطاف پذیری لیگاند و انعطاف پذیری گیرنده کار میکنند که در ادامه به طور مفصلتر درباره هر کدام صحبت کرده ایم.

انعطاف پذیری لیگاند
تغییر شکل لیگاند ممکن است در غیاب گیرنده ایجاد شده و متعاقباً متصل شود یا ممکن است هنگام پرواز در حضور حفره اتصال گیرنده، یا با انعطاف پذیری کامل چرخشی در هر قلاب دوضلعی با استفاده از اتصال قطعهای ایجاد شود. ارزیابی انرژی میدان نیرو اغلب برای انتخاب سازگاری منطقی از نظر انرژی مورد استفاده قرار میگیرد، اما روشهای دانش بنیان نیز استفاده شده است. پپتیدها مولکولهای بسیار انعطاف پذیر و نسبتاً بزرگ هستند، که انعطاف پذیری، مدل سازی آنها را به یک کار چالش برانگیز تبدیل میکند. تعدادی از روشها برای مدل سازی کارآمد انعطاف پذیری پپتیدها در هنگام اتصال پروتئین - پپتید توسعه داده شده است.
انعطاف پذیری گیرنده
ظرفیت محاسباتی در دهه گذشته به طرز چشمگیری افزایش یافته است و امکان استفاده از روشهای پیچیدهتر و محاسباتی بیشتر در طراحی داروها با رایانه را ممکن ساخته است. با این حال، برخورد با انعطاف پذیری گیرنده در روشهای داکینگ هنوز هم یک مسئله سخت است. دلیل اصلی این دشواری تعداد زیادی از درجههای آزادی است که باید در این نوع محاسبات در نظر گرفته شود. با این حال، غفلت از آن، در برخی موارد ممکن است منجر به نتایج ضعیف اتصال از نظر پیش بینی ژست اتصال شود. ساختارهای استاتیک متعددی که به صورت تجربی برای پروتئین یکسان در ترکیبات مختلف تعیین میشوند، اغلب برای شبیه سازی انعطاف پذیری گیرنده نیز مورد استفاده قرار میگیرند.
عملکرد امتیاز بندی
برنامههای داکینگ تعداد زیادی از لیگاندهای بالقوه را ایجاد میکند که برخی از آنها به دلیل برخورد با پروتئین بلافاصله رد میشوند. بقیه با استفاده از برخی از توابع نمره گذاری ارزیابی میشوند، که یک حالت را به عنوان ورودی میگیرد و عددی را برمیگرداند که نشان میدهد این ژست بیانگر یک تعامل اتصالی مطلوب است و یک لیگاند را نسبت به دیگری رتبه بندی میکند. بیشتر توابع نمره دهی، میدانهای مکانیکی مولکولی مبتنی بر فیزیک هستند که انرژی یک حالت قرارگیری خاص در محل اتصال را تخمین میزنند. مشارکتهای مختلف در اتصال را میتوان به عنوان یک معادله جمع نوشت:
اجزاء این معادله شامل اثرات حلال، تغییرات سازنده در پروتئین و لیگاند، انرژی آزاد به دلیل برهم کنش پروتئین و لیگاند، چرخشهای داخلی، انرژی پیوند لیگاند و گیرنده برای ایجاد یک انرژی واحد پیچیده و آزاد به دلیل تغییر در حالتهای ارتعاشی هستند. انرژی کم (منفی) نشان دهنده یک سیستم پایدار و در نتیجه یک فعل و انفعال احتمالی الزامی است. رویکردهای جایگزین از توابع نمره گذاری اصلاح شده استفاده میکند تا محدودیتهای بر اساس فعل و انفعالات کلیدی پروتئین - لیگاند یا پتانسیلهای مبتنی بر دانش ناشی از فعل و انفعالات مشاهده شده در پایگاههای بزرگ داده مربوط به ساختارهای لیگاند پروتئین (به عنوان مثال بانک داده پروتئین) را شامل شود. تعداد زیادی از ساختارهای کریستالوگرافی اشعه ایکس برای اجتماع بین پروتئینها و لیگاندهای با میل بالا اما تعداد نسبتاً کمتر برای لیگاندهای با میل کم وجود دارد، زیرا ترکیبات بعدی دارای ثبات کمتری هستند و بنابراین تبلور آنها دشوارتر است.
امتیازدهی توابع آموزش داده شده با این دادهها میتواند لیگاندهای با میل بالا را به طور صحیح متصل کند، اما همچنین برای لیگاندهایی که متصل نمیشوند، سازگاری متقابل منطقی ایجاد میکند. این تعداد زیادی از ضربههای مثبت کاذب را ایجاد میکند، به عنوان مثال، لیگاندهایی که پیش بینی میشود به پروتئین متصل میشوند که در صورت قرار گرفتن در یک لوله آزمایشی در واقع متصل نمیشوند. یکی از راههای کاهش تعداد موارد مثبت کاذب محاسبه مجدد انرژی موقعیتهای قرارگیری بهتر با استفاده از تکنیکهای دقیقتر اما دارای محاسبات بیشتر مانند روشهای Generalized Born یا پواسون - بولتزمن است.
انواع داکینگ مولکولی
همانطور که گفته شد تداخل بین پروتئینها و سایر مولکولها نقش مهمی در فرایندهای مختلف بیولوژیکی از جمله رونویسی و بیان ژن، تنظیم متابولیک، انتقال سیگنال و ارتباط سلولی ایفا میکند. آگاهی از جنبههای ساختاری یک پروتئین با شریک اتصال دهنده آن میتواند به درک مکانیسم چنین تعاملی کمک کند و بنابراین برای کشف و توسعه دارو مهم است. با این حال، بدست آوردن ساختارهای پیچیده با روشهای آزمایشی مانند کریستالوگرافی اشعه ایکس یا NMR همچنان مشکل و گران قیمت است. بنابراین، داکینگ مولکولی یک رویکرد مهم برای پیش بینی ساختارهای سه بعدی این قطعات مکمل در نظر گرفته میشود. انواع اتصالات مولکولهای مختلف با استفاده از داکینگ انجام میگیرند که در ادامه آنها را مورد بررسی قرار داده ایم.
داکینگ پروتئین - لیگاند چیست؟
در مورد «اتصال پروتئین - لیگاند» (Protein - Ligand Docking) میتوان گفت تعامل بین پروتئینها و لیگاندهای هم رده آنها نقش مهمی در بسیاری از فرایندهای بیولوژیکی ضروری و مسیرهای متابولیک از جمله هدایت سیگنال، نقل و انتقالات سلولی، تنظیم چرخه سلول، کنترل بیان ژن و مهار آنزیم دارد. بسیاری از تکنیکهای تجربی در حال حاضر برای تشخیص و اندازه گیری این فعل و انفعالات داکینگ در دسترس هستند، اما به دست آوردن ساختارهای پیچیده با روشهای آزمایشی مانند کریستالوگرافی اشعه ایکس یا NMR دشوار و گران قیمت است. بنابراین، داکینگ مولکولی یک رویکرد مهم برای مطالعه برهمکنش پروتئین - لیگاند و کشف و توسعه دارو در نظر گرفته میشود. امروزه از پیشرفته ترین ابزارهای نرم افزاری مربوط به لیگاند و پروتئین برای پیش بینی موقعیت و جهت یک لیگاند در زمان اتصال به گیرنده پروتئین با محاسبه مکان، هندسه و انرژی استفاده میشود.
فرایند داکینگ مولکولی یک لیگاند به جایگاه اتصال، از بر هم کنش طبیعی لیگاند و گیرنده آن از طریق کمترین مسیر انرژی تقلید میکند. به طور معمول روشهای مدل سازی با یک هدف از ساختار شناخته شده مانند ساختار کریستالوگرافی یک پروتئین مورد علاقه شروع میشوند. سپس از داکینگ برای پیش بینی اتصال کانفورماسیون و انرژی آزاد اتصال مولکولهای کوچک به هدف استفاده میشود. هر پروتکل اتصال را میتوان ترکیبی از یک الگوریتم جستجو و یک تابع نمره گذاری توصیف کرد. الگوریتم جستجو تعداد زیادی حالت از یک مولکول کوچک را در محل اتصال در نظر میگیرد و اجازه میدهد درجه آزادی سیستم پروتئین - لیگاند به اندازه کافی نمونه برداری شود تا حالتهای اتصال واقعی را شامل شود. تابع نمره گذاری نمره یا میل پیوندی یک ژست خاص که نشان دهنده ترمودینامیک فعل و انفعال سیستم پروتئین - لیگاند است را به منظور تشخیص حالتهای اتصال واقعی از سایر موارد مورد بررسی و رتبه بندی آنها بر این اساس محاسبه میکند.
آزمایشهای داکینگ تکی برای بررسی عملکرد پروتئین، مطالعه مهارکنندهها و بسترهای آنزیمی و روشن شدن مسیرهای بیوشیمیایی مفید هستند. مهمتر از همه، داکینگ میتواند برای غربالگری مجازی پایگاه دادههای بزرگ مواد شیمیایی موجود برای تشخیص و بهینه سازی مقادیر ترکیبات پیشرو دارویی استفاده شود، که فرصتهای بی نظیری را برای طراحی و کشف دارو بر اساس ساختار ارائه میدهد.

داکینگ پروتئین - پروتئین
در رابطه با «اتصال پروتئین - پروتئین» (Protein - Protein Docking) باید گفت برهم کنشهای پروتئین و پروتئین در جنبههای مختلف فرایندهای سلولی، از جمله کنترل متابولیک، انتقال سیگنال، تنظیم بیان ژن و ارتباطات سلولی، نقش اساسی دارند. مطالعات پروتئومیکس در سطح ژنوم، لیستی رو به افزایش از پروتئینهای متقابل را ارائه میدهد، اما تنها بخش کوچکی از کمپلکسهای بالقوه قابل تجزیه و تحلیل تجربی مستقیم است. دانستن جزئیات ساختاری یک برهمکنش پروتئین و پروتئین به ما کمک میکند تا مکانیسم تعاملهای اینچنین و در نتیجه عملکرد پروتئینهای شرکت کننده را درک کنیم. با این حال، بدست آوردن ساختارهای کریستالی مجتمعهای پروتئینی از پروتئینهای تکی دشوارتر است. علاوه بر این، بسیاری از فعل و انفعالات پروتئینی گذرا هستند، که مطالعه آنها را با کریستالوگرافی و NMR مشکل ساز میکند. بنابراین، نیاز به رویکردهای محاسباتی سریع و قوی برای پیش بینی قابل اعتماد ساختار مجتمعهای پروتئین و پروتئین در حال افزایش وجود دارد.
داکینگ پروتئین - پروتئین، پیش بینی محاسباتی ساختار پیچیده پروتئین با توجه به ساختار پروتئینی جزء جداگانه حل شده است، همانطور که در یک موجود زنده رخ میدهد. امروزه از پیشرفته ترین ابزارهای نرم افزاری برای پیدا کردن تغییر و ترکیب نسبی دو پروتئین استفاده میشود که منجر به یک مجموعه پایدار (از نظر انرژی مطلوب) خواهد شد. فرآیند داکینگ از دو مرحله اصلی تشکیل شده است: 1) ایجاد مجموعهای از پیکربندیها که به طور قابل اعتماد حداقل یک مورد تقریباً صحیح را شامل میشود و 2) تشخیص قابل اطمینان پیکربندیهای صحیح از سایر موارد با کمک عملکرد نمره گذاری. در روشهای جدید از جستجوی جهانی ساختار سفت و سخت بر اساس رویکرد همبستگی Fast Fourier Transform (FFT) شروع میشود که از فضای چرخشی و ترجمهای یک پروتئین در حالی که پروتئین دیگر را تثبیت میکند نمونه میگیرد.
اگر جایگاههای اتصال شناخته شده اند، میتوان از این اطلاعات برای کاهش فضای جستجو استفاده کرد. کانفورماسیونهای ایجاد شده سپس بر اساس شکل مکمل، الکترواستاتیک و امحاء درجه بندی و رتبه بندی شده و نتایج به دست آمده برای یافتن کانفورماسیونهای بسیار کم انرژی جمع آوری میشوند. ساختار (های) کمپلکس نماینده را میتوان با به حداقل رساندن انرژی غربال کرد. تکنیکهای اتصال محاسباتی برای جستجوی همه حالتهای اتصال احتمالی در فضای ترجمه و چرخش بین دو پروتئین استفاده میشود و هر ژست را با استفاده از یک تابع امتیازدهی مبتنی بر انرژی ارزیابی میکند. این یک وسیله مهم برای درک نیروهای فیزیک و شیمیایی است که زیربنای فعل و انفعالات ماکرومولکولی را تشکیل میدهند و یک ابزار ارزشمند برای مدل سازی ساختارهای پیچیده پروتئینی در سطح اتمی است. همچنین درک دقیق فعل و انفعالات پروتئین و پروتئین برای اهداف دخیل در بیماری برای طراحی منطقی داروهای مبتنی بر بیولوژی بسیار مهمتر است.

داکینگ پروتئین - پپتید
در رابطه با «اتصال پروتئین - پپتید» (Protein - Peptide Docking) میتوان گفت برهم کنش بین پروتئینها و پپتیدها نقش اساسی در ارگانیسمهای زنده ایفا میکند و میتوان آنها را در انواع مسیرهای سیگنالینگ که در محل سلولی، پاسخ ایمنی یا بیان و تخریب پروتئین نقش دارند، پیدا کرد. تداخلات نادرست پروتئین و پپتید با تعدادی از بیماریها (مانند سرطان، بیماریهای خود ایمنی) همراه است. درک تداخل پروتئین - پپتید برای طراحی دارویی هدفمند است و ویژگی ساختاری آنها موضوع داغ تحقیقات تجربی و نظری فعلی است. امروزه از پیشرفته ترین ابزارهای نرم افزاری برای مدل سازی برهمکنش پروتئین - پپتید و پیش بینی ساختار پیچیده استفاده میشود. روش کلی مدل سازی به دو مرحله تقسیم شده است: اول یک جستجوی کلی برای پیش بینی محل اتصال در سطح گیرنده پروتئین انجام میشود و در مرحله دوم یک روش اتصال محلی انعطاف پذیر برای مناسب سازی ساختار پپتید در محل اتصال شناخته شده استفاده میشود.
این دو مرحله تغییرات قابل توجهی را در هنگام اتصال دو جزء در نظر میگیرند. فرآیند تاشو پپتید به صراحت در پروتکل اتصال شبیه سازی شده است. هنگامی که دادههای تجربی یا پیش بینی شده در مورد بقایای محل اتصال در دسترس است، از چنین اطلاعاتی میتوان برای محدود کردن اتصال به مناطق محلی سطح پروتئین استفاده کرد. در نهایت، سازههای دارای نمره برتر، خوشه بندی شده، رتبه بندی شده و نمایندگان بزرگترین خوشهها قبل از ارائه برای اصلاح ساختاری با وضوح بالا انتخاب میشوند. امروزه از تکنیکهای اتصال محاسباتی برای جستجوی همه حالتهای اتصال احتمالی در فضای ترجمه و چرخشی بین گیرنده پروتئین و شریک اتصال دهنده پپتید استفاده میکند.
با توجه به ساختار گیرنده پروتئین و توالی پپتیدی، با استفاده از داکینگ مولکولی ساختار کمپلکس پروتئین - پپتید پیش بینی میشود که از ترکیبات و موقعیتهای تصادفی پپتید شروع میشود. این یک وسیله مهم برای درک نیروهای فیزیک و شیمیایی است که زیربنای فعل و انفعالات ماکرومولکولی را تشکیل میدهند و یک ابزار ارزشمند برای مدل سازی ساختارهای پیچیده پروتئین - پپتید در سطح اتمی است. علاوه بر این، درک دقیق این تعاملات، طراحی منطقی پپتیدهای بالقوه درمانی را تسهیل میکند.

داکینگ آنتی بادی - آنتی ژن
در مورد «اتصال آنتی بادی - آنتی ژن» (Antibody - Antigen Docking) میتوان گفت آنتی بادیها عناصر اصلی تشخیص سیستم ایمنی هستند که محکم و به طور خاص به آنتی ژنهای مختلف متصل میشوند تا فعالیت آنها را مسدود کرده یا آنها را برای نابودی مشخص کنند. آنتی بادیها به طور فزایندهای به عنوان ابزارهای تشخیصی و داروهای درمانی مورد استفاده قرار میگیرند و یکی از اجزای مهم فرایند طراحی و مهندسی آنتی بادی، در دسترس بودن ساختار سه بعدی با کیفیت بالا از مجتمعهای آنتی بادی و آنتی ژن است. کریستالوگرافی اشعه ایکس دقیق ترین و صحیح ترین اطلاعات را در مورد ساختار پروتئین و فعل و انفعالات آن ارائه میدهد، اما کریستالیزاسیون مجتمعهای آنتی بادی و آنتی ژن یک چالش عمده است و تعداد ساختارهای آنتی بادی تعیین شده در مجتمع با آنتی ژنهای آنها هنوز نسبتاً کم است. در مقابل، مدل سازی محاسباتی یک مسیر سریع و ارزان را برای ایجاد مدلهای ساختاری برای چنین مجموعه هایی فراهم میکند.
دومینهای متغیر (Fvs) زنجیره سنگین و سبک مورد توجه ویژهای در مدل سازی محاسباتی هستند، زیرا معمولاً بیشتر یا همه ویژگی یک آنتی بادی را برای هدف آنتی ژن خود نشان میدهند. امروزه پروتکل مدل سازی پیشرفته، برهم کنشهای آنتی بادی و آنتی ژن و همچنین انعطاف پذیری مطلوب منطقه CDR (به ویژه حلقه H3) را در نظر میگیرد و از جدیدترین ابزارهای نرم افزاری داکینگ برای یافتن تغییر و ترکیب نسبی آنتی بادی و آنتی ژن دخیل در تشکیل کمپلکس با انرژی مطلوب استفاده میکند. روشهای کلی عبارتند از: اول، یک جستجوی ساختار سفت و سخت انجام میشود و مدلهای پیچیده آنتی بادی - آنتی ژنی از نظر هندسی قابل قبول تولید میشوند. در این فرایند محدودیتهای تعریف شده توسط کاربر (به دست آمده از دادههای تجربی) میتواند برای محدود کردن فضای جستجو اعمال شود.
مدلهای بدست آمده با استفاده از یک تابع نمره گذاری، که بطور خاص برای سیستمهای آنتی بادی توسعه یافته است، نمره گذاری و رتبه بندی میشوند. سپس سازههای دارای بهترین نمره دستهبندی میشوند و نمایندگان بزرگترین دستهها برای بهینه سازی ساختاری با کم کردن انرژی قبل از ارائه انتخاب میشوند. امروزه از تکنیکهای داکینگ محاسباتی برای جستجوی همه حالتهای اتصال احتمالی در فضای ترجمه و چرخش بین یک آنتی بادی (Fv ، Fab یا آنتی بادی کامل) و آنتی ژن آن با مدل سازی تغییرات تطبیقی حلقههای متغیر بر روی تشکیل کمپلکس استفاده میشود. این یک وسیله مهم برای درک نیروهای فیزیک و شیمیایی است که زیربنای فعل و انفعالات ماکرومولکولی را تشکیل میدهند و یک فرضیه کار بر اساس مجموعه کمپلکس ایجاد میشود و هدایت آزمایشهای بیشتر برای طراحی منطقی درمانهای مبتنی بر آنتی بادی را انجام دهد.

داکینگ پروتئین - اسید نوکلئیک
در مورد «اتصال پروتئین - اسید نوکلئیک» (Protein - Nucleic Acid Docking) باید گفت که فعل و انفعالات بین پروتئین و اسیدهای نوکلئیک در فرآیندهای مختلف بیولوژیکی ضروری، از جمله تکثیر DNA، رونویسی RNA، پیوند RNA، تخریب اسیدهای نوکلئیک و سنتز پروتئین، نقش اساسی دارند. نقص در برهم کنش پروتئین و اسید نوکلئیک در تعدادی از بیماریها از اختلالات عصبی گرفته تا سرطان دخیل است. درک ما از این فرآیندها با حل شدن ساختارهای جدید مجتمعهای پروتئین - اسید نوکلئیک و تجزیه و تحلیل جزئیات ساختاری فعل و انفعالات بهبود مییابد. با این حال، تعیین تجربی اکثر ساختارهای پیچیده پروتئین - اسید نوکلئیک با روشهای با وضوح بالا یک فرایند خسته کننده و دشوار است. تکنیکهای محاسباتی مکمل رویکردهای تجربی در روشن کردن برهمکنشهای پروتئین و اسید نوکلئیک است.
در داکینگ In Silico پروتئینها و اسیدهای نوکلئیک، با ایجاد مدلهای نظری ساختارهای پیچیده در جزئیات اتمی، میتواند اطلاعات کافی برای ایجاد یک فرضیه و راهنمایی تجزیه و تحلیلهای تجربی بیشتر برای شناسایی اسیدهای آمینه مهم یا بقایای نوکلئوتیدها را ارائه دهد. امروزه از پیشرفته ترین نرم افزارهای محاسباتی برای پیدا کردن تغییر و ترکیب نسبی پروتئین و اسید نوکلئیک دخیل در تشکیل کمپلکس با انرژی مطلوب استفاده میشود. روشهای کلی عبارتند از: اول، یک جستجوی جامع ساختار سخت انجام میشود و ساختارهای پیچیده پروتئین - اسید نوکلئیک قابل قبول از نظر هندسی ایجاد میشوند. در این فرایند میتوان محدودیتهای تعریف شده توسط کاربر را برای محدود کردن فضای جستجو اعمال کرد. مدلهای بدست آمده با استفاده از پتانسیلهای آماری، به طور خاص برای مجموعههای پروتئین - RNA یا پروتئین - DNA، نمره گذاری و رتبه بندی میشوند.
سپس سازههای دارای بهترین نمره خوشه بندی میشوند و نمایندگان بزرگترین خوشهها برای بهینه سازی ساختاری با کم کردن انرژی قبل از ارائه انتخاب میشوند. در روشهای جدید داکینگ از تکنیکهای محاسباتی برای جستجوی همه حالتهای اتصال احتمالی در فضای ترجمه و چرخش بین پروتئین و اسیدهای نوکلئیک (DNA ، RNA یا DNA/RNA ترکیبی) استفاده میشود که میتواند بر اساس شکل گیری پیچیده تغییر شکل بسیار زیادی را متحمل شود. این یک وسیله مهم برای درک نیروهای فیزیکوشیمیایی است که زیربنای فعل و انفعالات ماکرومولکولی را تشکیل میدهند و یک ابزار ارزشمند برای مدل سازی ساختارهای پیچیده پروتئین - اسید نوکلئیک در سطح اتمی است. علاوه بر این، درک دقیق این فعل و انفعالات شامل اهداف دخیل در بیماری برای طراحی منطقی درمانهای بیولوژیکی بسیار مهمتر است.

داکینگ پروتئین - لیپید
در رابطه با «اتصال پروتئین - لیپید» (Protein - Lipid Docking) میتوان گفت که پروتئینهای غشایی نقش مهمی در طیف وسیعی از عملکردهای سلولی ایفا میکنند. ثابت شده است که اتصال پروتئینها به غشاهای بیولوژیکی، سیگنال دهی سلولی و بسیاری دیگر از رویدادهای مهم سلولی را از طریق مکانیسمهای مختلف تنظیم میکند. برهم کنشهای درون بدن با لیپیدهای غشایی به تثبیت ساختار این پروتئینها و ترویج ترتیب مجدد، مونتاژ، تفکیک یا تغییرات ساختاری در بسیاری از حوزههای ساختاری پروتئین، که منجر به فعال شدن فعالیت بیولوژیکی آنها میشود، کمک میکند. برخی از پروتئینهای سیتوزولیک از طریق حوزههای ساختاری خاص غشای سلولی جذب میشوند. علاوه بر این، چربیها نیز بسترهای طبیعی بسیاری از آنزیمهای مهم در مسیرهای متابولیکی کلیدی هستند. بنابراین، درک تعامل بین پروتئینها و چربیها از اهمیت بسیار بالایی برخوردار است.
تداخل پروتئین - لیپید با تشکیل پیوندهای هیدروژنی بین مولکولی، فعل و انفعالات واندروالس، برهم کنشهای آبگریز و پلهای یونی (به ویژه باقی ماندههای آسپارتات یا گلوتامات) بین پروتئین و لیگاند چربی تثبیت میشود. از نرم افزارهای محاسباتی پیشرفته برای پیش بینی این برهمکنشهای پروتئین و لیپید استفاده میشود. محل اتصال چربی یک پروتئین را میتوان از توالی اسید آمینه آن یا از ساختار سه بعدی آن با استفاده از پروتکلهای اتصال مولکولی پیش بینی کرد. برخی از روشهای داکینگ اطلاعات توالی و ساختار را ترکیب کرده و از نظر انرژی مطلوب ترین مجموعه پروتئین - لیپید را پیدا میکنند. همچنین پایداری مجموعههای متصل را میتوان از طریق شبیه سازیهای دینامیک مولکولی بیشتر آزمایش کرد.

داکینگ پروتئین - کربوهیدرات
در مورد «اتصال پروتئین - کربوهیدرات» (Protein - Carbohydrate Docking) باید گفت کربوهیدراتها در بسیاری از مسیرهای مهم سیگنالینگ نقش دارند و تشخیص کربوهیدراتها جزء کلیدی بسیاری از فرایندهای بیولوژیکی از جمله ایمنی ذاتی، متابولیسم و پاسخ ایمنی است. به عنوان مثال، گلیکانهایی که روی سطوح سلولی یا مولکولهای بیولوژیکی ترشح شده قرار دارند، نقش مهمی در برهمکنش سلول - سلول از جمله برهمکنش بین سلول میزبان و عوامل بیماریزا ایفا میکنند. اهمیت فیزیولوژیکی و پاتولوژیکی فعل و انفعالات گلیکان - پروتئین توجه زیادی را در زمینه طراحی دارو بر اساس ساختار جلب میکند. با این حال، مطالعه این تعاملات با کریستالوگرافی اشعه ایکس بسیار دشوار بوده است، زیرا بخشهای قند معمولاً دارای ساختار و ترکیب شیمیایی ناهمگن هستند. تکنیکهای مدل سازی مولکولی محاسباتی یک روش جایگزین جذاب برای مطالعه تشخیص و ویژگی پروتئین - کربوهیدرات ارائه میدهند.
در داکینگ مولکولی این فعل و انفعالات پروتئین - کربوهیدرات از طریق ترکیب یک الگوریتم جستجوی سازگار ژنتیکی همراه با یک تابع انرژی آزاد تجربی خاص برای کربوهیدراتها پیش بینی میشوند. روش داکینگ به شما امکان میدهد کاوش سیستماتیک جهتها و موقعیتهای لیگاند کربوهیدرات را در حفره پروتئین (که در مقایسه با محل اتصال مولکولهای کوچک معمولاً بازتر است و به خوبی تعریف نشده است) انجام دهید و انرژیهای آزاد کمپلکسهای پیچیده را ارزیابی میکند. محدودیتهای مشتق شده از آزمایش نیز میتوانند برای پروتکل اتصال به منظور محدود کردن فضای جستجو برای قند و همچنین مولکول پروتئین اعمال شوند. عملکرد انرژی به طور خاص برای کربوهیدراتها طراحی شده است، که برای خوشه بندی حالتهای اتصال و اکتشاف سازههای پیچیده با انرژی مناسب استفاده میشود. به علاوه پایداری نسبی مجتمعهای لیگاند - پروتئینی را میتوان با اجرای شبیه سازی دینامیک مولکولی کوتاه ارزیابی کرد.
با توجه به اهمیت روزافزون پروتئینهای متصل کننده کربوهیدرات به عنوان اهداف دارویی احتمالی، روش مدل سازی محاسباتی که برخی از ویژگیهای مولکولی متمایز منحصر به فرد برای مجتمعهای پروتئین - کربوهیدرات را آشکار میکند برای ارائه درک سطح اتمی از تشخیص کربوهیدرات مناسب است. رویکردهای جدید داکینگ میتواند تعامل بین پروتئینها و لیگاندهای کربوهیدراتهای جداگانه را پیش بینی کند و همچنین توانایی پیش بینی حالتهای اتصال و پیوندهای گلیکان را با پروتئینهای متصل کننده به گلیکان دارد.

دقت داکینگ مولکولی چقدر است؟
دامنه و قابلیتهای داکینگ در طول سالها مورد بحث قرار گرفته است. تا به امروز، چندین مطالعه معیار در مقایسه برنامههای مختلف داکینگ منتشر شده است. به طور متوسط، نشان داده شده است که دقت روشهای داکینگ در شناسایی موقعیتهای صحیح در حدود 60 تا 75 درصد است. یک چالش عمده همچنان پیش بینی دقیق تعامل انرژی بین دو مولکول است. تا کنون، همبستگیهای کمی بین فعالیتهای آزمایشی و امتیازات داکینگ به طور کلی، به دلیل سطح تقریبی بالای اعمال شده در توابع امتیازدهی، کم است. با این حال، ارتباطات کیفی کاملاً قابل قبول هستند که با موفقیت چندین کمپین غربالگری مجازی مبتنی بر داکینگ ثابت شده است.
اغلب اتفاق میافتد که حالت قرارگیری با امتیاز بالاتر لزوماً بهترین حالت نیست. به مثال زیر توجه کنید: «برومو دومینها» (Bromodomains) پروتئینهای کوچکی با نقوش محافظت شده بوده که مسئول بیان یا سرکوب ژن هستند. یک مهار کننده بسیار قوی برومودومین BET مهارکننده IBET762 است، یک لیگاند «تریازولازپین» (Triazolazepine) که دارای IC50 در محدوده نانومولار است. با استفاده از Vina برای ذخیره مجدد با استفاده از PDB ID 3P5O ، بهترین ژست در جهت کاملاً متفاوتی نسبت به مرجع کریستالوگرافی قرار دارد. مسئله دیگر عملکرد در «اتصال متقاطع» (Cross-docking) است: متصل کردن لیگاندهای مرجع به ساختارهای غیر بومی همان پروتئین اغلب منجر به پیش بینی اشتباه حالت داکینگ میشود. استراتژیهای مختلفی برای افزایش دقت اتصال استفاده شده است. دو رویکرد اصلی عبارتند از Ensemble docking و Consensus docking که در دو زیربخش بعدی مورد بحث قرار میگیرد.

داکینگ گروهی
یکی از کاربردهای اولیه «داکینگ گروهی» (Ensemble docking) توسط Kuntz و Oshiro منتشر شد. داکینگ گروهی با انجام شبیه سازیهای متعدد اتصال بر روی پروتئینهای مختلف انجام میشود، این روش تا حدی انعطاف پذیری پروتئین را در بر میگیرد. فرض اصلی این است که اگر یک لیگاند داده شده در ساختار بومی امتیاز خوبی بگیرد، این لیگاند باید همین کار را در برابر گروهی از کنفورماسیونها انجام دهد که امکان روشن شدن بهتر تعاملات محافظت شده را فراهم میآورند. به این ترتیب، داکینگ گروهی میتواند به عنوان داکینگ چهار بعدی (4D) در نظر گرفته شود و میزان موفقیت آن ۷۸ درصد از صحت هندسه اتصال گزارش شده است. مجموعهای از سازگاری مولکول مورد نظر ممکن است از دادههای کریستالوگرافی، NMR یا مدلهای محاسباتی، به عنوان مثال، دینامیک مولکولی یا مدلهای همولوژیکی مختلف ناشی شود.
مواردی که ممکن است بر عملکرد داکینگ گروهی تاثیر بگذارند شامل نحوه امتیازدهی، ساخت گیرندههای گروه، انعطاف پذیری لیگاند، شباهت مولکولی و غنی سازی هستند. اتصال گروهی همچنین به عنوان تکنیکی مداخله گر برای شبیه سازیهای پیچیدهتر عمل میکند. با استفاده از دادههای سه بعدی (3D) میتوان تقریبی عمیقتر از تناسب القایی، که ممکن است با روشهای دیگر مانند QSAR ادغام شود، انجام دهید. اتصال گروهی در طیف گستردهای از کاربردها مانند شناسایی نقاط داغ برای تعامل پروتئین - پروتئین، مدل سازی GPCR، مدل سازی ساختارهای apo و holo، «شناسایی برخورد» (hit identification)، شناسایی تعدیل کنندههای گیرنده هستهای، متابولیسم فاز I و بررسی سمیت موفقیت آمیز بوده است.

داکینگ عام
همانطور که در مقدمه توضیح داده شد، توصیه میشود از بیش از یک برنامه معتبر برای غربالگری مجازی استفاده کنید. با توجه به دامنههای مختلف توابع نمره گذاری و الگوریتمها، انتظار میرود که آنها مکمل یکدیگر باشند. با این حال، از آنجا که توابع نمره گذاری با مبانی مختلف ساخته میشوند، مقایسه و ترکیب آنها ساده نیست. اولین تلاش توسط Charifson Corkery انجام شد، در آن مطالعه، نویسندگان با استفاده از سه عملکرد نمره دهی مستقل (CHEMSCORE ، PLP و DOCK) بهبود مداوم را نشان دادند.
مقالات بعدی نشان داد که ترکیب سه تا چهار تابع نمره گذاری مستقل برای بهبود نتایج کافی است. یک روش جایگزین ایجاد توابع امتیازدهی جدید بود که روشهای موجود را ترکیب میکرد: نمونههای برتر DrugScore و X-Score هستند. در حالی که این استراتژی در موارد خاص مفید بود، هنوز قدرت پیش بینی کمتر از میانگین نسبت به لیگاندهای بسیار انعطاف پذیر دارد. مطمئناً امتیازدهی گروهی، انتخاب ژست را بهبود میبخشد اما به طور مستقیم به نمرات ترکیب شده بستگی دارد، به عنوان مثال، یک همبستگی قوی بین آنها ممکن است میزان خطا را افزایش دهد.
علاوه بر این، توابع نمره دهی نسبت به ویژگیهای خاص جایگاههای اتصال مناسب هستند. رویکردهای جایگزین برای داکینگ گروهی مبتنی بر رتبه و تقاطع هستند که از معیارهای آماری برای ارزیابی سهم و اهمیت موضعهای الزام آور استفاده میکنند. این روشها باعث غنی سازی و رتبه بندی بهتر میشوند و دارای مزیتی هستند که نیازی به ورودی یا محاسبه دیگر فراتر از آمار ندارند. کاربردهای موفقیت آمیز این روش شامل شناسایی آنزیم ترانسکریپتاز معکوس ویروس HIV، مهار کنندههای توبولین و ویروس زیکا هستند.
رویکردهای داکینگ مولکولی
دو رویکرد به ویژه در انواع اتصال مولکولی رایج است. یک رویکرد از «تکنیک تطبیق» (Matching Technique) استفاده میکند که پروتئین و لیگاند را به عنوان سطوح مکمل توصیف میکند. روش دوم «فرآیند اتصال واقعی» (Actual Docking Process) را شبیه سازی میکند که در آن انرژی برهم کنش زوج لیگاند و پروتئین محاسبه میشود. هر دو رویکرد دارای مزایای قابل توجهی و همچنین برخی محدودیتها هستند. این موارد در ادامه بیان شده است.
روش شکل مکمل
روشهای تطابق هندسی یا «مکمل بودن شکل» (Shape complementarity)، پروتئین و لیگاند را به عنوان مجموعهای از ویژگیها توصیف میکند که باعث اتصال آنها میشود. این ویژگیها ممکن است شامل توصیف کنندههای سطح مولکولی یا سطح مکمل باشد. در این مورد، سطح مولکولی گیرنده از نظر «سطح قابل دسترسی به حلال» (Solvent - Accessible Surface Area) و سطح مولکولی لیگاند از نظر توصیف سطح مطابقت آن توصیف میشود. «مکمل بودن» (The complementarity) بین دو سطح به توصیف انطباق آنها میپردازد که ممکن است به یافتن حالت مکمل اتصال به هدف و مولکولهای لیگاند کمک کند.

رویکرد دیگر، توصیف ویژگیهای آبگریز پروتئین با استفاده از چرخش در اتمهای زنجیره اصلی است. روش دیگر استفاده از تکنیک توصیف شکل «فوریر» (Fourier) است. در حالی که رویکردهای مبتنی بر مکمل بودن شکل معمولاً سریع و قوی هستند آنها معمولاً نمیتوانند حرکات یا تغییرات پویا در ترکیب لیگاند - پروتئین را به طور دقیق مدل کنند، اگرچه پیشرفتهای اخیر به این روشها اجازه میدهد تا انعطاف پذیری لیگاند را بررسی کنند. روشهای مکمل بودن شکل میتواند چندین هزار لیگاند را در عرض چند ثانیه اسکن کرده و در واقع بفهمد که آیا میتوانند در محل فعال پروتئین متصل شوند و معمولاً حتی برای برهمکنش پروتئین - پروتئین مقیاس پذیر است. آنها همچنین بسیار بیشتر در معرض رویکردهای مبتنی بر «فارماکوفور» (داروبر) هستند، زیرا از توصیف هندسی لیگاندها برای یافتن اتصال بهینه استفاده میکنند.
شبیه سازی
شبیه سازیِ فرایند اتصال بسیار پیچیدهتر است. در این رویکرد، پروتئین و لیگاند با فاصله فیزیکی از هم جدا میشوند و لیگاند پس از تعداد مشخصی حرکت در فضای تطبیقی، موقعیت خود را در محل فعال پروتئین مییابد. این حرکتها ساختارهای سخت مانند برگردان و چرخش را یکی کرده و همچنین تغییرات داخلی در ساختار لیگاند از جمله چرخش زاویه دورانی ایجاد میکند. هر یک از این حرکتها در فضای ترکیب لیگاند باعث ایجاد کل هزینه انرژی سیستم میشود، بنابراین کل انرژی سیستم پس از هر حرکت محاسبه میشود.
مزیت آشکار شبیه سازی اتصال این است که انعطاف پذیری لیگاند به راحتی در آن گنجانده میشود، در حالی که تکنیکهای مکمل بودن شکل باید از روشهای مبتکرانه برای انعطاف پذیری در لیگاندها استفاده کنند. همچنین، واقعیت را دقیقتر مدل سازی میکند، در حالی که تکنیکهای مکمل بودن شکل، بیشتر انتزاعی هستند. بدیهی است که شبیه سازی از نظر محاسباتی گران قیمت است و باید چشم انداز وسیعی از انرژی را کاوش کرد. تکنیکهای مبتنی بر شبکه، روشهای بهینه سازی و افزایش سرعت کامپیوتر، شبیه سازی داکینگ را واقعیتر کرده است.

انواع حالت های داکینگ
انعطاف پذیری پروتئین و لیگاند و ارزیابی اتصال یکی از چالشهای اصلی در داکینگ است. مسلماً یک رویکرد مناسب برای آزمایش رفتار کمپلکس پروتئین - لیگاند در یک محیط پویا قرار دارد. از زمان معرفی مدل امیل - فیشر برای اتصال، با ابزارها و روشهای کارآمدتر مشخص شد که استفاده از دستکاری پروتئینها راه بهتری است. به هر حال، مشکلات لیگاند یکی از مسائل اصلی در شیمی دارویی و داروسازی بوده است. با رشد تصاعدی مخازن مانند «بانک داده پروتئین» (PDB) الگوهای تا شونده جدید و انواع ترتیب ساختاری کشف شد. این اطلاعات به شما امکان میدهد الگوها و شباهتهایی را در محلهای اتصال و پاکتهای پروتئینی مشاهده کنید تا عملکرد داخلی پروتئینها را روشن کند.
داکینگ گیرنده سفت و لیگاند سفت
هنگامی که لیگاند و گیرنده هر دو به عنوان اجسام سفت و سخت رفتار میکنند، با توجه به تنها سه درجه آزادی ترجمه و سه درجه چرخش، فضای جستجو بسیار محدود است. در این مورد، انعطاف پذیری لیگاند را میتوان با استفاده از مجموعهای از ترکیبات لیگاند از پیش محاسبه شده تا درجاتی از همپوشانی اتم - اتم بین پروتئین و لیگاند مورد استفاده قرار داد. نسخههای اولیه DOCK ، FLOG و برخی برنامههای اتصال پروتئین و پروتئین، مانند FTDOCK، از روشی استفاده کردند که لیگاند و گیرنده را در طول فرآیند سفت نگه میداشت. DOCK اولین روش خودکار برای اتصال یک مولکول به محل گیرنده است و به طور مداوم در حال توسعه است.
این روش، لیگاند و گیرنده را به عنوان مجموعهای از کرهها توصیف میکند که میتوانند با استفاده از روش تشخیص گروهی روی هم قرار گیرند. همچنین از الگوریتمهای تطبیق هندسی و شیمیایی استفاده میشود و مجموعههای لیگاند - گیرنده را میتوان با درنظر گرفتن تناسب استریک، مکمل شیمیایی یا شباهت فارماکوفور نمره گذاری کرد. در نسخههای بهبود یافته آن، روش ساخت افزایشی و جستجوی جامع برای در نظر گرفتن انعطاف پذیری لیگاند اضافه میشوند. جستجوی جامع به طور تصادفی تعدادی از موارد سازگار تعریف شده توسط کاربر را به عنوان مضرب تعداد پیوندهای قابل چرخش در لیگاند تولید میکند.
FLOG سازگاری لیگاند را بر اساس هندسه فاصله ایجاد میکند و از الگوریتم پیدا کردن گروه برای محاسبه مجموعه فاصلهها استفاده میکند و تا تعداد 25 تطابق صریح لیگاند میتواند برای انعطاف پذیری مورد استفاده قرار گیرد. FLOG به کاربران اجازه میدهد تا نقاط ضروری که باید با اتم لیگاند جفت شوند را تعریف کنند. این رویکرد در صورتی مفید است که از پیش، تعامل مهمی قبل از اتصال شناخته شده باشد. کانفورماسیونها با توجه به واندروالس، الکترواستاتیک، پیوند هیدروژنی و فعل و انفعالات آبگریز، با یک تابع نمره گذاری میشوند.
داکینگ لیگاند انعطاف پذیر و گیرنده سفت
برای سیستمهایی که رفتار آنها از الگوی تناسب القایی پیروی میکند اهمیت انعطاف پذیری لیگاند و گیرنده بسیار مهم است، زیرا در آن صورت هر دو لیگاند و گیرنده تغییر شکل میدهند تا حداقل مجموعه کاملاً مناسب انرژی را تشکیل دهند. با این حال، وقتی گیرنده نیز انعطاف پذیر است هزینه بسیار زیاد است. بنابراین رویکرد رایج، همچنین یک معامله بین دقت و زمان محاسبه، این است که با لیگاند انعطاف پذیر رفتار میشود در حالی که گیرنده در هنگام اتصال محکم نگه داشته میشود. تقریباً همه برنامههای داکینگ مانند AutoDock و FlexX از این روش استفاده کرده اند. AutoDock 3.0 از روشهای الگوریتم ژنتیک لمارکی، تکاملی و انلینگ شبیه سازی شده مونت کارلو برای مدلسازی انعطاف پذیری لیگاند در عین سفت نگه داشتن گیرنده استفاده میکند.
عملکرد نمره گذاری بر اساس میدان نیروی AMBER، از جمله وان دروالس، پیوند هیدروژنی، فعل و انفعالات الکترواستاتیک، آنتروپی تطبیقی و شرایط انحلال است. هر عبارت با استفاده از یک عامل مقیاس تجربی بدست آمده از دادههای تجربی وزن میشود. AutoDock 4.0 با حرکت زنجیرهای جانبی میتواند انعطاف پذیری گیرنده را مدل کند. علاوه بر این، تعامل اتصال پروتئین - پروتئین را میتوان در این نسخه از AutoDock ارزیابی کرد. AutoDock Vina به تازگی به عنوان آخرین نسخه برای اتصال مولکولی و غربالگری مجازی منتشر شده است. با جمع آوری مجدد 190 مجتمع گیرنده - لیگاند که به عنوان مجموعه آموزشی AutoDock 4 استفاده میشد، AutoDock Vina به طور همزمان تقریباً دو برابر پیشرفت نمایی در سرعت و دقت قابل توجهی در پیش بینی حالت اتصال را نشان داد.
FlexX از الگوریتم ساخت افزایشی برای نمونه برداری از لیگاند استفاده میکند. قطعه پایه ابتدا با تطبیق جفتهای پیوند هیدروژنی و فعل و انفعالات حلقه آروماتیک بین لیگاند و پروتئین در محل فعال متصل میشود. سپس اجزای باقیمانده مطابق با مجموعهای از زاویههای پیچشی از پیش تعیین شده برای انعطاف پذیری لیگاندها ساخته میشوند. عملکرد نمره گذاری FlexX بر اساس کار Bohm است. نسخه فعلی آن شامل شرایط فعل و انفعالات الکترواستاتیک، پیوندهای هیدروژنی جهت دار، آنتروپی چرخشی و برهمکنشهای آروماتیک و چربی دوست است. تعاملات بین گروههای عملکردی نیز با تعیین نوع و هندسه برای گروهها در نظر گرفته میشود.
داکینگ گیرنده انعطاف پذیر و لیگاند انعطاف پذیر
ثابت شده است که تحرک ذاتی پروتئینها ارتباط تنگاتنگی با رفتار اتصال به لیگاند دارد و توسط Teague بررسی شده است. گنجاندن انعطاف پذیری گیرنده یک چالش مهم در زمینه داکینگ است. در حالت ایده آل، با استفاده از شبیه سازی دینامیک مولکولی (MD) میتوان تمام درجات آزادی را در مجموعه گیرنده - لیگاند مدل کرد. اما شبیه سازی دینامیک مولکولی مشکل نمونه گیری ناکافی را دارد که قبلاً به آن اشاره کردیم. عامل دیگر هزینه محاسباتی بالای آن است که مانع استفاده از این روش در غربالگری پایگاه دادههای شیمیایی بزرگ میشود.
علاوه بر تناسب القایی تاریخی چندین مدل نظری، انتخاب سازگار و القاء سازهای، برای نشان دادن روند اتصال لیگاند - پروتئین انعطاف پذیر پیشنهاد شده است. با توجه به تعریفی که تیگ ارائه داده است، انتخاب سازگار به فرایندی اطلاق میشود که یک لیگاند به طور انتخابی به کانفورماسیون مطلوب از تعدادی از ترکیبات پروتئینی متصل میشود، القاء کانفورماسیونی فرایندی را توصیف میکند که در آن لیگاند پروتئین را به شکل دیگری تبدیل میکند که خود به خود در حالت عادی آن را انجام نمیدهد. در برخی موارد، این تبدیل کانفورماسیون را میتوان به تا شدن نسبی پروتئین تشبیه کرد. در ادامه انواع روشها برای داکینگ گیرنده انعطاف پذیر و لیگاند انعطاف پذیر بررسی شده اند.
داکینگ نرم
در حال حاضر روشهای مختلفی برای اعمال انعطاف پذیری گیرنده در دسترس است. ساده ترین آن به اصطلاح «داکینگ نرم» (soft-docking) است که انرژی دافعه وان در والس را در عملکرد نمره گذاری کاهش میدهد تا امکان همپوشانی اتم - اتم بین گیرنده و لیگاند را فراهم آورد. به عنوان مثال، پتانسیل LJ 8-4 در GOLD و پتانسیل صاف در AutoDock 3.0 متعلق به این کلاس است. این روش ممکن است شامل انعطاف پذیری کافی نباشد. با این وجود، این مزیت بهره وری محاسباتی دارد زیرا مختصات گیرنده به سادگی با تنظیم پارامترهای وان دروالس تنظیم میشوند.
استفاده از کتابخانه روتامر
استفاده از کتابخانههای روتامر روش دیگری برای مدل سازی انعطاف پذیری گیرنده است. کتابخانههای روتامر شامل مجموعهای از کانفورماسیونهای زنجیره جانبی هستند که معمولاً از تجزیه و تحلیل آماری دادههای تجربی ساختاری تعیین میشوند. مزیت استفاده از روتامرها سرعت نسبی در نمونه برداری و پرهیز از کوچک شماری موانع است. ICM (مکانیک مختصات داخلی) برنامهای است که از کتابخانههای روتامر با متدولوژی احتمال جانبدارانه استفاده میکند، همراه با Monte Carlo کانفورماسیون لیگاند را جستجو میکند.
انعطاف پذیری زنجیره جانبی گیرنده
AutoDock 4 از یک روش نمونهگیری همزمان برای مقابله با انعطاف پذیری زنجیره جانبی استفاده میکند. چندین زنجیره جانبی گیرنده میتواند توسط کاربران انتخاب شده و همزمان با یک لیگاند با استفاده از روشهای مشابه نمونه برداری شود. سایر قسمتهای گیرنده در طول نمونه برداری با نقشه انرژی شبکه به طور سخت تلقی میشوند. نقشه انرژی شبکه معرفی شده توسط Goodford برای ذخیره اطلاعات انرژی گیرنده و ساده سازی محاسبه انرژی متقابل بین لیگاند و گیرنده استفاده میشود.
کانفورماسیون های گروهی پروتئین
روش دیگر برای سر و کار داشتن با انعطاف پذیری پروتئین، استفاده از مجموعهای از ترکیبات پروتئینی است که مطابق با نظریه انتخاب سازگار است. یک لیگاند به طور جداگانه به مجموعهای از ترکیبات پروتئینی سفت و محکم متصل میشود و نتایج بسته به روش انتخابی ادغام میشوند. این روش در ابتدا در DOCK اجرا شد، که به طور متوسط شبکه انرژی بالقوه گروهی را ایجاد میکند و در بسیاری از برنامهها به روشهای مختلف گسترش مییابد. به عنوان مثال، FlexE ساختارهای کریستالی متعددی از یک پروتئین خاص را جمع آوری کرده و قسمتهای مشابه را ادغام میکند در حالی که مناطق متفاوت را به عنوان جایگزینهای مختلف علامت گذاری میکند. در طول ساخت افزایشی یک لیگاند از پروتئینهای گسسته به شکل ترکیبی نمونه برداری میشود. ساختار پروتئینی با بالاترین امتیاز بر اساس مقایسه بین لیگاند و هر جایگزین انتخاب میشود. این روشها به طور خلاصه در جدول زیر بررسی شده اند.
| روش | توضیحات | مزایا | معایب | نرم افزار (های) مورد استفاده |
| «پتانسیل نرم» (Soft potential) | vdW را تغییر میدهد تا امکان همپوشانی بین اتمهای گیرنده و لیگاند وجود داشته باشد. | کارایی محاسباتی
پیاده سازی آسان استفاده ترکیبی با روشهای دیگر | انعطاف پذیری نامناسب
انعطافپذیری را به صورت ضمنی، نامناسب و به روش غیر کمی شرح میدهد. | GOLD
AutoDock |
| «کتابخانه روتامر» (Rotamer library) | کتابخانه زنجیره جانبی برای دستیابی به کانفورماسیونهای ممکن را جستجو میکند. | کارایی محاسباتی نسبی
پرهیز از کوچک کردن موانع | وابستگی شدید به پایگاه داده استفاده شده
عدم انعطافپذیری زنجیره اصلی | ICM |
| انعطاف پذیری زنجیره جانبی گیرنده | زنجیره جانبی و کنفورماسیوهای لیگاند را به طور همزمان با استفاده از GA نمونه گیری میکند. | کارایی محاسباتی نسبی
مدلسازی اثر لیگاند روی رزیجوهای جایگاه اتصال | تنها درگیری زنجیرههای جانبی انتخابی
عدم انعطافپذیری زنجیره اصلی | AutoDock 4 |
| کانفورماسیونهای گروهی پروتئین | اتصال لیگاند به یک سری از ساختارهای گیرنده که نشان دهنده حالتهای مختلف کانفورماسیونی است. | شامل انعطافپذیری کامل و واضح | هزینه محاسباتی گرانقیمت
محدود به کانفورماسیونهای پروتئینی مورد استفاده در نمونهگیری | DOCK
FlexE |
روش هیبرید یا ترکیبی
روش ترکیبی یکی دیگر از استراتژیهای عملی برای مدل سازی انعطاف پذیری گیرنده است. یک مثال خوب Glide است، یک برنامه بسیار محبوب در زمینه داکینگ به شمار میرود. Glide یک سری فیلترهای ترتیبی را طراحی میکند تا حالتها و جهتهای احتمالی لیگاند را در محل اتصال گیرنده جستجو کند. انعطاف پذیری لیگاند با جستجوی جامع در فضای زاویه چرخش لیگاند انجام میشود. کانفورماسیونهای اولیه لیگاند بر اساس انرژی پیچشی انتخاب شده و در مکانهای اتصال گیرنده با عوامل باالقوه نرم متصل میشود. سپس از یک شناسایی روتامر برای مدل سازی انعطاف پذیری گیرنده استفاده میشود. IFREDA از یک روش ترکیبی استفاده میکند که مجموعهای از عوامل باالقوه نرم و ترکیب گیرندههای متعدد را در بر میگیرد و انعطاف پذیری گیرنده را محاسبه میکند. برنامههای دیگر، مانند QXP و Affinity، جستجوی مونت کارلو در زمینه کانفورماسیون لیگاندها را با کم کردن مراحل انجام کار دنبال میکنند.
در حین کم کردن مراحل، قسمتهای تعریف شده از پروتئین مجاز توسط کاربر قابل حرکت هستند تا از برخورد اتم بین لیگاند و گیرنده جلوگیری شود. SLIDE به گونهای طراحی شده است که انعطاف پذیری را با قابلیت حذف برخوردها (چرخش پیوند منفرد هر یک از لیگاندها یا زنجیره های جانبی پروتئین) ترکیب میکند. یک روش بهینه سازی بر اساس «نظریه میدان میانگین» (Mean - field theory) برای مدل سازی مکملهای القایی مناسب بین لیگاند و پروتئین استفاده میشود.
روشهای ذکر شده در بالا فقط شامل انعطاف پذیری زنجیره جانبی یا انعطاف پذیری کامل گیرنده است. ما میدانیم که لوپهای تشکیل دهنده جایگاههای فعال نقش مهمی در اتصال لیگاند دارند. در برخی موارد حلقه یا لوپ ممکن است تغییر شکل چشمگیری داشته باشد در حالی که در قسمتهای دیگر گیرنده تغییر چندانی در اتصال لیگاند ایجاد نمیشود. برای این وضعیت، روشهای انعطاف پذیری زنجیره جانبی در نمونه برداری از پروتئین مناسب شکست میخورند و به نظر میرسد در نظر گرفتن انعطاف پذیری کامل هدر دادن محاسبات است. در تصویر زیر به عنوان مثال ساختارهای کریستالی روی هم قرار داده شده را نشان میدهد. جایگاه فعال تریوز فسفات ایزومراز دارای یک حلقه 11 اسیدآمینهای است که ۷ انگستروم بالای با اتصال لیگاند حرکت میکند. با این حال، بقیه آنزیمها در مقایسه با ساختارهای آپو (غیر وابسته به لیگاند) و هولو هیچ حرکتی ندارند.

چندین خانواده آنزیمی نیز شامل تنظیم مجدد حلقه در جایگاه فعال مسئول اتصال لیگاندها، مانند Bromodomain یک خانواده گسترده مربوط به اتصال استیل - لیزین یا دی هیدروفولات ردوکتاز، مسئول نگهداری تجمعات سلولی تتراهیدروفولات و سایر انواع کینازها هستند. علاوه بر اینها روش نمونه گیری حلقه Local Move Monte Carlo (LMMC) یک رویکرد جدید است که بر نمونه برداری از ترکیب لیگاند در جایگاههای فعال حاوی حلقه تمرکز میکند.
ارزیابی داکینگ مولکولی
وابستگی متقابل بین نمونه برداری و عملکرد نمره گذاری بر قابلیت اتصال در پیش بینی حالتهای محتمل یا نزدیکی الزامی برای ترکیبات جدید تأثیر میگذارد. بنابراین، ارزیابی یک پروتکل اتصال به طور کلی (هنگامی که دادههای تجربی در دسترس است) برای تعیین قابلیت پیش بینی آن مورد نیاز است. ارزیابی اتصال میتواند با استفاده از استراتژیهای مختلف انجام شود، مانند:
- محاسبه دقت اتصال (DA)
- ارتباط بین نمره اتصال و پاسخ تجربی یا تعیین عامل غنی سازی (EF)
- فاصله بین بخش اتصال دهنده یون - یون در جایگاه فعال آنزیم
- وجود مدلهای القایی مناسب
در ادامه هر کدام از این موارد توضیح داده شده اند.
دقت اتصال
«دقت اتصال در داکینگ مولکولی» (Docking accuracy) نشان دهنده یک معیار برای تعیین کمیت تناسب برنامه اتصال با توجیه توانایی پیش بینی موقعیت مناسب لیگاند با توجه به مشاهده تجربی است.
عامل غنی سازی
صفحههای اتصال نیز میتوانند با غنی سازی لیگاندهای حاشیهای اتصال دهندههای شناخته شده از بین یک پایگاه داده بزرگ از مولکولهای فریبنده غیرقابل اتصال فرضی ارزیابی شوند. به این ترتیب، موفقیت یک صفحه اتصال با ظرفیت آن برای غنی سازی تعداد کمی از ترکیبات فعال شناخته شده در ردههای بالای صفحه از بین تعداد بسیار بیشتری مولکول فرضی در پایگاه داده ارزیابی میشود. سطح زیر منحنی ویژگی عملکرد گیرنده (ROC) به طور گستردهای برای ارزیابی عملکرد آن مورد استفاده قرار میگیرد.
آینده نگری
ضربههای ناشی از صفحههای اتصال به اعتبار دارویی (به عنوان مثال IC50، اندازه گیری میل یا قدرت) میرسد. فقط مطالعات آینده نگر اثبات قطعی مناسب بودن تکنیک برای یک هدف خاص است.
معیارسازی
پتانسیل برنامههای اتصال را برای بازتولید حالتهای اتصال دهنده که توسط کریستالوگرافی اشعه ایکس تعیین میشود میتوان با طیف وسیعی از مجموعههای معیار اتصال بارگذاری کرد. برای مولکولهای کوچک، چندین مجموعه داده معیار برای اتصال و غربالگری مجازی وجود دارد. مجموعه متنوع Astex متشکل از ساختارهای کریستالی اشعه ایکس پروتئین - لیگاند با کیفیت بالا یا فهرست «ابزارهای مفید» (Directory of Useful Decoys) یا DUD برای ارزیابی عملکرد غربالگری مجازی است. ارزیابی برنامههای اتصال به دلیل پتانسیل آنها برای بازتولید حالتهای اتصال پپتید را میتوان با درسهای ارزیابی کارآیی اتصال و امتیازدهی (LEADS - PEP) ارزیابی کرد.
کاربرد داکینگ مولکولی
تعامل اتصال بین یک لیگاند مولکول کوچک و یک پروتئین آنزیمی ممکن است منجر به فعال شدن یا مهار آنزیم شود. اگر پروتئین گیرنده باشد، اتصال لیگاند ممکن است منجر به آگونیسم (اتصال یک ماده شیمیایی به گیرنده و فعال کردن پاسخ بیولوژیک) یا آنتاگونیسم (اتصال یک ماده شیمیایی به گیرنده و جلوگیری از فعالیت آن) شود. داکینگ بیشتر در زمینه طراحی دارو مورد استفاده قرار میگیرد، اکثر داروها مولکولهای ارگانیک کوچکی هستند و داکینگ مولکولی ممکن است به دلایل زیر انجام شود:
- شناسایی برخورد، داکینگ به همراه یک تابع نمره گذاری میتواند برای غربالگری سریع پایگاههای اطلاعاتی بزرگ داروهای بالقوه در رایانه (In Silico) برای شناسایی مولکولهایی که احتمالاً به هدف مورد علاقه پروتئین متصل میشوند استفاده شود که به آن «غربالگری مجازی» (Virtual Screening) نیز گفته میشود.
- بهینه سازی مقادیر ترکیبات پیشرو دارویی، اتصال یا داکینگ میتواند برای پیش بینی اینکه در کدام جهت و در کدام جهت نسبی یک لیگاند به پروتئین متصل میشود (همچنین به عنوان حالت اتصال یا ژست نامیده میشود) استفاده شود. این اطلاعات ممکن است به نوبه خود برای طراحی آنالوگهای قویتر و انتخابیتر مورد استفاده قرار گیرد.
- «زیست پالایی» (Bioremediation)، داکینگ لیگاند - پروتئین همچنین میتواند برای پیش بینی آلایندههایی که توسط آنزیمها تجزیه میشوند، مورد استفاده قرار گیرد.
داکینگ مولکولی داروها

فعل و انفعالات بین مولکولهای زیستی برای همه فرایندهای بیولوژیکی ضروری است. با استفاده از این فعل و انفعالات، موجودات زنده شبکههای متقابل نظارتی و متابولیکی پیچیدهای را حفظ میکنند که با هم فرایندهای حیات را تشکیل میدهند. درک پیچیدگی مسیرهای بیولوژیکی و فعل و انفعالات بین ماکرومولکولهای درگیر ضروری شده است و داکینگ مولکولی به طور جامع به این نیاز پاسخ داده است. اتصال مولکولی ممکن است به عنوان یک مفهوم شیمی محاسباتی تعریف شود که راهحلهایی را برای کشف مکانیزم تعامل بستر و لیگاند ارائه میدهد. این روش مولکول تعاملی را بر اساس توپوگرافی آنها در کنار هم قرار میدهد. هدف اصلی داکینگ مولکولی تعیین بهترین ترکیب ممکن پروتئین - لیگاند، پروتئین - پروتئین یا سایر انواع فعل و انفعالات با حداقل انرژی است.

داکینگ همچنین به عنوان یک تکنیک حیاتی در مدل سازی برهمکنش پروتئین - لیگاند و پروتئین - پروتئین در نظر گرفته میشود و در نتیجه در مطالعات مربوط به رمزگشایی عملکرد مولکولی مجموعههای مختلف شرکت میکند. رویکرد داکینگ مولکولی یک حالت قدرتمند برای مدل سازی برهمکنش پروتئین و مولکولهای کوچک در سطح اتمی است. این روش بسیار در توصیف رفتار مولکولهای کوچک در محل اتصال آنها کمک میکند. دو مرحله اصلی که در داکینگ متصل میشوند عبارتند از: (1) بدست آوردن ترکیب لیگاند پایدار و (2) ارزیابی میل اتصال آن.
در اکثر موارد، محلهای اتصال قبل از انجام داکینگ پیش بینی میشوند. مکانهای اتصال معمولاً با مقایسه هدف مورد نظر با پروتئینهای دیگر از همان خانواده با عملکرد مشابه بدست میآیند. کاربردهای داکینگ گسترده هستند، اطلاعات به دست آمده عمیق است و مسیر را برای استفاده از داکینگ کاملاً باز کرده است تا پژوهشگران وارد عرصه جدیدی از تحقیقات با تمرکز بر طراحی مولکولی، کشف هدف و تجزیه و تحلیل پتانسیل کاربرد ترکیبات شوند. معمولاً اولین قدم برای یافتن ترکیبات فعال از مواد شیمیایی موجود برای پروژه کشف و توسعه دارو است.
اگرچه بسیاری از شرکتهای دارویی منابع آنلاین و کتابخانههای خود را دارند که ممکن است میلیونها ترکیب داشته باشند، نگهداری کتابخانه و انجام غربالگری با توان بالا هزینهبر است. غربالگری مجازی یک روش جایگزین برای انجام غربالگری میلیونها ترکیب در عرض چند روز ارائه میدهد. داکینگ مولکولی یکی از کاربردی ترین روشهای غربالگری مجازی است، به ویژه هنگامی که ساختار سه بعدی پروتئین مورد نظر در دسترس باشد. در واقع، اتصال مولکولی بیش از سه دهه است که استفاده میشود و تعداد زیادی داروی جدید کشف شده و بر این اساس توسعه یافته است. اگرچه شکی نیست که این روش نقش مهمی را ایفا خواهد کرد، اما اتصال مولکولی از نظر میزان موفقیت هنوز با موفقیت کامل فاصله دارد.
FBDD یا طراحی دارو بر اساس قطعات به عنوان یک الگوی موفق برای توسعه داروهای جدید پدیدار شد و آمیزهای از غربالگری توان بالا «مبتنی بر هدف» (Target - based) و رویکردهای طراحی دارو بر اساس ساختار را ترکیب کرد. داکینگ مولکولی، از برنامه ریزی و اولویت بندی ترکیب کتابخانه قطعه گرفته تا یافتن آنالوگهایی با احتمال اتصال بهتر از طریق غربالگری مجازی در مقیاس بزرگ کتابخانههای مرکب kra مهمی در FBDD دارد. ML یا Machine Learning شاخهای از هوش مصنوعی است که در زمینههای مختلف علم و فناوری بسیار مورد توجه قرار گرفته است و روشهای اتصال مولکولی نیز از این منطقه پویا استفاده میکنند. اگرچه اخیراً، انعطاف پذیری Machine Learning در مدل سازی دادهها توابع امتیاز دهی متنوعتر و دقیق تری را به طور ضمنی برای ویژگیهای بیشتر مجموعه پیچیده در نظر گرفته است.
محدودیت های داکینگ مولکولی
محدودیت اصلی داکینگ مولکولی به دلیل عدم اطمینان در توانایی نمره گذاری توابع برای ایجاد انرژی اتصال صحیح است. این امر ناشی از این واقعیت است که برخی از فعل و انفعالات بین مولکولی مانند اثر حلالی و تغییر آنتروپی به سختی به طور دقیق پیش بینی میشوند. علاوه بر این، برخی از واکنشهای بین مولکولی که اهمیت آنها ثابت شده است به ندرت در توابع نمره دهی در نظر گرفته میشوند. به عنوان مثال، پیوند هالوژن تأیید میشود که در میل به اتصال پروتئین - لیگاند و تعاملات گوانیدین - آرژنین نیز موثر است، اما در داکینگ در نظر گرفته نمیشود.
کمپلکس ترانستیرتین - تیروکسین، یکی از نمونههای مهمی است که در آن توابع انرژی شکست میخورند. شبیه سازی اتصال با توابع انرژی منجر به تولید دو حالت اتصال شد یکی شبیه به حالت اتصال بومی تیروکسین و دیگری متعلق به یک حوزه اتصال متناوب با میانگین انحراف مربع ریشه (RMSD) 8/97 Å از حالت اتصال بومی. شبیه سازی انرژی انجام شد و راه حل با انرژی کمتر که توسط برنامه داکینگ انتخاب شد، همان راه حل با RMSD بالاتر بود. بنابراین، در این مورد داکینگ مولکولی نتوانست پیش بینی صحیح حالت اتصال را انجام دهد. بنابراین ممکن است پاسخهای منفی کاذب در طول برنامه رخ دهند.
برخورد دقیق با مولکولهای آب در محل متصل کننده در حین داکینگ، هنوز یک مشکل حل نشده است، که کار سختی است و به دو دلیل به توجه زیادی در آینده نزدیک نیاز دارد. اولاً، ساختارهای بلوری اشعه ایکس به دلیل پراکندگی ناکارآمد توسط اتمهای کوچکتر فاقد اطلاعات مختصری از هیدروژن هستند. عدم آگاهی از موقعیت دقیق هیدروژن منجر به عدم دقت در شناسایی مولکولهای آب میشود که ممکن است به عنوان یک مولکول پل بین لیگاند و گیرنده عمل کنند. ثانیاً، هیچ رویکرد نظری قابل اعتمادی برای پیش بینی دقیق چگونگی تأثیر مولکولهای آب توسط لیگاندها و میزان تأثیر آن در دسترس نیست. علاوه بر این، با دانش فعلی ما نمیتوان پیش بینی کرد که چند مولکول آب در محل اتصال با لیگاندهای احتمالی جایگزین میشود و چگونه شبکه پیوند هیدروژنی با اتصال لیگاند مختل میشود.
یکی از چالشهای عمده دیگری که در زمینه داکینگ با آن روبرو هستیم وجود گیرنده سفت و سخت است. یک پروتئین بسته به لیگاندی که به آن متصل میشود، میتواند شکلهای متفاوتی را به خود بگیرد. در نتیجه، اتصال با استفاده از یک گیرنده سفت و سخت با یک ترکیب گیرنده مطابقت دارد که منجر به منفی کاذب در بسیاری از موارد میشود که بعداً لیگاند فعال شناخته شد. این امر به این دلیل اتفاق میافتد که یک پروتئین میتواند در حال حرکت مداوم بین حالتهای مختلف سازهای با انرژی مشابه وجود داشته باشد، که معمولاً در اتصال به آن نادیده گرفته میشود. سرانجام، طیف فعالیت در برابر پروتئینهای خارج از هدف چیزی است که حتی در صفحههای محاسباتی به ندرت دیده میشود و فقط توسط آزمایشات انسانی و حیوانی مورد بررسی قرار میگیرد.




با سلام و وقت بخیر. بسیار عالی و اموزنده بود. یک راهنمایی می خواستم. لایسنس نرم افزار لیگ پلات من تمام شده و برای دریافت نسخه ی جدید، ایمیل آکادمیک مانع دریافت ایمیل حاوی لایسنس می شود. برای دانلود این نرم افزار روش دیگری وجود دارد؟ یا نرم افزار مشابهی؟ متشکرم.
دستتون درد نکنه خیلی مطلب مفیدی بود
سلام، وقت شما بخیر،
از اینکه مطلب برای شما مفید واقع شده بسیار خرسندیم.
از همراهی شما با فرادرس سپاسگزاریم.