نرم افزارهای بیوانفورماتیک کاربردی در ژنتیک | معرفی مهارت های نرم افزاری ضروری
نرم افزارهای بیوانفورماتیک ابزارهای کامپیوتری هستند که دقت و سرعت اجرای فعالیتهای آزمایشگاهی و پروژههای تحقیقاتی را افزایش میدهند. در این مقاله، به معرفی مهمترین نرم افزارهای بیوانفورماتیک و وب سایتهای کاربردی مورد استفاده در آن میپردازیم.
اهمیت یادگیری نرم افزارهای بیوانفورماتیک در تحقیقات پزشکی و زیست شناسی چیست؟
بیوانفورماتیک، به عنوان یک رشته جدید در حال ظهور، ریاضیات، فناوری اطلاعات و زیست شناسی را ترکیب کرده و به پاسخگویی به سوالات بیولوژیکی کمک میکند. کلمه «بیوانفورماتیک» برای اولین بار در سال 1968 استفاده شد و تعریف آن برای اولین بار در سال 1978 ارائه شد. بیوانفورماتیک همچنین به عنوان زیست شناسی محاسباتی نامیده شده است. اجزای اصلی بیوانفورماتیک عبارتند از توسعه ابزارها و الگوریتمهای نرم افزاری و تجزیه و تحلیل و تفسیر دادههای بیولوژیکی با استفاده از انواع ابزارهای نرم افزاری و الگوریتمهای خاص.


بیوانفورماتیک به عنوان شاخهای بین رشتهای از علوم زیستی، توسعه روشهای شناختی، تجزیه و تحلیل برای کشف مقادیر وسیع پایگاه دادههای بیولوژیکی جهت پشتیبانی از ذخیره سازی، ترتیب، سیستم بندی، درک و اجرای دادههای بیولوژیکی و به عنوان یک وسیله مورد آزمایش برای تحقیقات ژنوم و محصول ژنتیکی استفاده میشود. این ابزارها شامل انفورماتیک سنتی، مدرن و محاسبات ابری، آمار و ریاضیات و تشخیص الگو، بازسازی، یادگیری ماشینی، شبیه سازی، مدل سازی مولکولی و الگوریتمهای مربوط به فولدینگ ساختارها است. با این حال، رشد و پیشرفت حوزه دانش بیولوژیکی ارتباط تنگاتنگی با نرم افزار و برنامه نویسی کامپیوتری دارد و برای مدیریت مقادیر زیادی از توالیهای مولکولی DNA ، RNA، پروتئینها، متابولیتها و تجزیه و تحلیل ساختاری و عملکردی مورد نیاز است. نرم افزارهای بیوانفورماتیک در زمینههای مختلف علوم مورد استفاده قرار میگیرند. چند حوزه مهم در زیر آورده شده است:
- پزشکی مولکولی
- پزشکی فرد محور
- داروهای پیشگیرانه
- ژن درمانی
- توسعه دارو
- کاربردهای ژنوم میکروبی
- پاکسازی زباله
- مطالعات تغییرات اقلیمی
- منابع انرژی جایگزین
- بیوتکنولوژی
- مقاومت آنتی بیوتیکی
- تجزیه و تحلیل میکروبها در پزشکی قانونی
- ایجاد سلاحهای زیستی
- مطالعات تکاملی
- بهبود محصول
- مقاومت در برابر حشرات
- بهبود کیفیت تغذیه
- توسعه انواع گیاهان مقاوم به خشکی
- علم دامپزشکی
نرم افزارهای بیوانفورماتیک ابزارهایی هستند که برای استخراج اطلاعات معنی دار از پایگاههای داده زیست شناسی مولکولی - بیولوژیکی و انجام توالی یابی یا تجزیه و تحلیل ساختاری طراحی شده اند. عواملی که هنگام طراحی ابزارها، نرم افزارها و برنامههای بیوانفورماتیک باید مورد توجه قرار گیرند عبارتند از: اولاً ممکن است کاربر نهایی (زیست شناس) کاربر مداوم فناوری رایانه نباشد پس باید کاربرپسند باشند، دوم این ابزارهای نرم افزاری باید با توجه به توزیع جهانی جامعه تحقیقات علمی از طریق اینترنت در دسترس همگان قرار گیرند.
وب سایت ها و نرم افزارهای بیوانفورماتیک مهم
مرکز ملی اطلاعات بیوتکنولوژی (NCBI 2001) بیوانفورماتیک را اینگونه تعریف میکند: بیوانفورماتیک زمینهای از علم است که در آن زیست شناسی، علوم رایانه و فناوری اطلاعات در یک رشته واحد ادغام میشوند. سه زیر شاخه مهم در زمینه بیوانفورماتیک وجود دارند (1) توسعه الگوریتمها و آمار جدید که با آنها روابط بین اعضای مجموعه دادههای بزرگ را ارزیابی میکند (۲) تجزیه و تحلیل و تفسیر انواع مختلف دادهها از جمله توالی نوکلئوتیدها و اسیدهای آمینه، حوزههای پروتئینی و ساختارهای پروتئینی (۳) توسعه و پیاده سازی ابزارهایی که دسترسی کارآمد و مدیریت انواع مختلف اطلاعات را ممکن میسازند. به طور خاص، این علم توسعه پایگاههای داده و الگوریتمهای رایانهای برای تسهیل و تسریع تحقیقات بیولوژیکی است. همچنین از آن برای شناسایی اهداف مولکولی جدید برای کشف دارو استفاده میشود. در ادامه پایگاههای داده بیولوژیکی و ابزارها و نرم افزارهای بیوانفورماتیکی را معرفی میکنیم.
پایگاه های اطلاعاتی بیولوژیکی
پایگاههای اطلاعاتی بیولوژیکی کتابخانههای اطلاعات علوم زیستی هستند که از آزمایشات علمی، مقالات منتشر شده، فناوری آزمایش با توان بالا و تجزیه و تحلیل محاسباتی جمع آوری شده است. آنها حاوی اطلاعاتی از مناطق تحقیقاتی از جمله ژنومیکس، پروتئومیکس، متابولومیکس، «بیان ژن ریزآرایه» (microarray gene expression) و فیلوژنتیک هستند. اطلاعات موجود در پایگاههای داده بیولوژیکی شامل عملکرد ژن، ساختار، محل (هر دو سلولی و کروموزومی)، اثرات بالینی جهشها و همچنین شباهت توالیها و ساختارهای بیولوژیکی است.

مفاهیم پایگاه داده رابطهای علوم رایانه و مفاهیم بازیابی اطلاعات کتابخانههای دیجیتال برای درک پایگاههای اطلاعاتی بیولوژیکی مهم هستند. طراحی پایگاه داده بیولوژیکی، توسعه و مدیریت بلند مدت یکی از زمینههای اصلی رشته بیوانفورماتیک است.
محتویات دادهها شامل توالی ژن، توصیف متنی، ویژگیها و طبقه بندی موجودات، استنادها و دادههای جدول است. این موارد اغلب به عنوان دادههای نیمه ساختار یافته توصیف میشوند و میتوانند به صورت جداول، سوابق کلیدی و ساختارهای XML ارائه شوند. ارجاع متقابل بین پایگاههای داده با استفاده از شمارههای دسترسی به پایگاه داده رایج است. پایگاههای اطلاعاتی بیولوژیکی یک ابزار مهم برای کمک به دانشمندان در درک و توضیح مجموعهای از پدیدههای بیولوژیکی از ساختار مولکولهای زیستی و برهم کنش آنها برای کل متابولیسم موجودات زنده و درک تکامل گونهها است.

این دانش به تسهیل مبارزه با بیماریها کمک میکند و همچنین به توسعه داروها و کشف روابط اساسی بین گونهها در تاریخ حیات کمک میکند. دانش بیولوژیکی بین پایگاههای اطلاعاتی عمومی و تخصصی مختلف توزیع میشود.این امر گاهی اوقات تضمین ثبات اطلاعات را دشوار میکند. پایگاههای اطلاعاتی بیولوژیکی از پایگاههای داده دیگر با شمارههای پیوست به عنوان یکی از راههای پیوند دادن دانش مربوط به یکدیگر استفاده میکنند. دو دسته برای پایگاههای اطلاعاتی بیولوژیکی در بیوانفورماتیک وجود دارد، اول پایگاه داده نوکلئوتیدی و دوم پایگاه داده پروتئین.
- پایگاه دادههای نوکلئوتیدی توسط مرکز ملی اطلاعات بیوتکنولوژی (NCBI) (http://www.ncbi.nlm.nih.gov.in) و آزمایشگاه زیست شناسی مولکولی اروپا (EMBL) (http://www.ebi.ac.uk) و DNA DataBank ژاپن (DDBJ) (http://www.ddbj.nig.ac.jp) توسعه یافته است.
- پایگاه دادههای پروتئینی عبارتند از منابع اطلاعات پروتئین (PIR) (http://pir.georgetown.edu) ، SWISS-PROT (http://www.expasy.ch/sprot/) و بانک اطلاعات پروتئین (PDB) (http: //www.pdb.org). علاوه بر این موارد پایگاههای داده پروتئینی ثانویه دیگری مانند Pfam ، PRINTS ، BLOCKS ، PROSITE ، InterPro و غیره نیز وجود دارد.
انواع نرم افزارهای بیوانفورماتیک کدام ها هستند؟
ابزارهای بیوانفورماتیک برنامههای نرم افزاری برای ذخیره، بازیابی و تجزیه و تحلیل دادههای بیولوژیکی و استخراج اطلاعات از آنها هستند. عواملی که هنگام طراحی این ابزارها باید مورد توجه قرار گیرند عبارتند از: توجه به کاربر نهایی (زیست شناس) ممکن است کاربر به صورت مکرر یک استفاده کننده از فناوریهای رایانهای نباشد و بنابراین باید نرم افزار بیوانفورماتیک مورد نظر بسیار کاربر پسند باشد و مورد دوم اینکه این ابزارهای نرم افزاری باید با توجه به توزیع جهانی جامعه تحقیقات علمی از طریق اینترنت در دسترس قرار گیرند. به صورت کلی محصولات استاندارد و سفارشی وجود دارند که نیازهای یک پروژه خاص را برآورده میکنند.
نرم افزارهای داده کاوی وجود دارند که دادهها را از پایگاههای داده توالی ژنومی بازیابی میکنند و همچنین ابزارهای تجسم برای تجزیه و تحلیل و بازیابی اطلاعات از پایگاههای داده پروتئومی وجود دارد. نرم فزارهای بیوانفورماتیک را میتوان به موارد زیر طبقه بندی کرد:
- دستههای اصلی ابزارهای بیوانفورماتیک
- ابزارهای بررسی همولوژی و شباهت
- تجزیه و تحلیل توالی
- نرم افزارهای بیوانفورماتیک تجزیه و تحلیل عملکرد پروتئین
- نرم افزارهای بیوانفورماتیک تجزیه و تحلیل ساختاری
در ادامه هر کدام از این گروه نرم افزارهای بیوانفورماتیک را به صورت جداگانه بررسی میکنیم.
دسته های اصلی ابزارهای بیوانفورماتیکی
نرم افزارهای بیوانفورماتیک استاندارد و سفارشی وجود دارند که نیازهای پروژههای خاص را برآورده میکنند. نرم افزارهای داده کاوی وجود دارند که دادهها را از پایگاههای دادههای توالی ژنومی بازیابی میکنند و همچنین ابزارهای تجسم برای تجزیه و تحلیل و بازیابی اطلاعات از پایگاههای داده پروتئومی وجود دارند. این نرم افزارهای بیوانفورماتیکی را میتوان به عنوان ابزارهای همولوژی و شباهت، ابزارهای تجزیه و تحلیل پروتئین، ابزارهای تجزیه و تحلیل توالی و ابزارهای مختلف طبقه بندی کرد. در ادامه برخی از نرم افزارهای مهم در حوزه بیوانفوراتیک را بررسی میکنیم.
نرم افزار بیوانفورماتیک Vector NTI چیست و چه کاربردی دارد؟
نرم افزار Invitrogen Vector NTI مجموعهای کاملاً یکپارچه از ابزارهای تجزیه و تحلیل و طراحی توالی است. این نرم افزار از شما در مدیریت، نمایش، تجزیه و تحلیل، تبدیل، به اشتراک گذاری و انتشار انواع مختلف دادههای بیولوژیکی مولکولی در یک محیط تجزیه و تحلیل با کیفیت بالا پشتیبانی میکند. به عنوان مثال، به محققان اجازه میدهد تا قبل از انجام آزمایش در آزمایشگاه، آزمایش شبیه سازی DNA را بر روی کامپیوتر برنامه ریزی کنند. این برنامه دارای امکاناتی است که در ادامه به آنها اشاره کرده ایم.
- ایجاد، حاشیه نویسی، تجزیه و تحلیل و به اشتراک گذاری توالی DNA یا پروتئین
- انجام و ذخیره جستجوهای BLAST
- طراحی پرایمرها برای PCR، شبیه سازی، تعیین توالی یا آزمایش هیبریداسیون
- برنامه ریزی انجام کلونینگ و ران کردن ژل در محیط In - Silico
- ترازبندی توالی چند پروتئین یا DNA
- جستجو در NCRE's Entrez و مشاهده و ذخیره پروتئینها
- ویرایش دادههای کروماتوگرام، جمع آوری آنها
با استفاده از ابزار Vector NTI data export میتوانید دادههای مولکولی خود را از قالبهای فایل Vector NTI دارای حق چاپ به فرمتهای استاندارد منبع باز صادر کنید. نرم افزار Vector NTI Express Designer یک راه حل پیشگامانه برای اجرای فنی شبیه سازی و مونتاژ In - Silico نسل بعدی است. رابط کاربری که به تازگی توسعه یافته است، یک جدول ساخت مجازی را برای ساختارهای وکتور کشیدن و رها کردن ساده و مقایسه توالی ساده بین پروژهها ارائه میدهد. نرم افزار Vector NTI Express Designer همچنین دسترسی به ابزارهای وب Invitrogen GeneArt برای دستیابی به سطوح بهینه بیان پروتئین را تسهیل میکند. نرم افزار بیوانفورماتیک Vector NTI Express Designer اکنون برای سیستم عاملهای Mac یا PC در دسترس است.

نرم افزار GALAXY
Galaxy یک پلت فرم باز و مبتنی بر وب در دسترس و یکی از نرم افزارهای بیوانفورماتیک قابل تولید و شفاف برای تحقیقات محاسباتی است. GALAXY یک نرمافزار محبوب است که به طور گستردهای در یکپارچه سازی دادهها، آنالیز و ماندگاری دادهها برای زیست شناسی محاسباتی در کنار یک سستم عامل مانند UNIX در دسترس افراد است. این ابزار بیوانفورماتیک دارای مزایای زیادی است که در ادامه به آنها میپردازیم.

- قابل دسترس بودن: تجربه برنامه نویسی برای بارگذاری آسان دادهها، اجرای ابزارهای پیچیده و گردش کار و تجسم نتایج، در استفاده از این نرم افزار مورد نیاز نیست.
- قابل تکرار بودن: GALAXY اطلاعات را ضبط میکند تا کاربر مجبور نباشد هر دفعه آنها را ذخیره کند. هر کاربری میتواند یک تحلیل محاسباتی کامل از پارامترهای ابزاری گرفته تا «درخت وابستگی» (dependency tree) را تکرار و درک کند.
- شفاف سازی: کاربران این پلتفرم سابقه، گردش کار و تجسمات خود را از طریق وب به اشتراک میگذارند و منتشر میکنند.
- جامعه محور بودن: کاربران فراگیر و متنوع (توسعه دهندگان، مربیان، محققان، پزشکان و غیره) دارای اختیار هستند که یافتههای خود را به اشتراک بگذارند.
کاربردهای GALAXY شامل مطالعه در بیان ژن، پروتئومیکس، ترنسکریپتومیکس، «آنالیز توالی یابی نسل بعدی» (NGS) و مونتاژ ژنوم است.

Chromas چیست؟
Chromas یک نمایشگر و ویرایشگر رایگان، ساده و کاربر - پسند برای استفاده جهت کروماتوگرامهای حاصل از توالییابی سنگر است. این برنامه دارای گزینههای تبدیل فرمت بسیاری از جمله توابع پردازش دستهای برای مدیریت همزمان بسیاری از فایلها است. Chromas دارای ویژگیهای بسیاری است از جمله اینکه انواع فایلهای کروماتوگرام با فرمت SCF و ZTR را باز میکند که توسط ترتیب دهندههای دیگر ایجاد شده یا از پایگاه دادهها بازیابی شده اند. قابلیت چاپ کروماتوگرام را با گزینههای بزرگنمایی یا متناسب با یک صفحه میدهد. توالیهای با کیفیت پایین یا توالی وکتورها را در صورت موجود بودن دادههای با کیفیت حذف میکند.
توالیها را در متن ساده مانند FASTA ، FASTQ ، EMBL ، GenBank یا فرمتهای GCG خروجی میدهد یا به صورت بازهای شمارهگذاری شده آنها را ارائه میدهد. توالی را در کلیپ بورد با متن ساده، قالب FASTA یا FASTQ برای چسباندن به سایر برنامهها کپی میکند. توالی و کروماتوگرام را برعکس و مکمل میکند، زیر توالیها را با مطابقت دقیق یا ترازبندی بهینه جستجو میکند. ترجمهها را در 3 فریم همراه با توالی نمایش میدهد، میتوانید به کمک آن یک تصویر از بخش کروماتوگرام را برای چسباندن در اسناد یا ارائهها کپی کنید. ChromasPro برای مونتاژ توالی خوانده شده در کانتیگها، همراه با ویرایشگر پیکربندی گرافیکی است که کروماتوگرامهای تراز را نمایش می دهد.
AutoDimer چیست و چه کاربردی دارد؟
AutoDimer یکی از نرم افزارهای بیوانفورماتیک است که برای غربالگری سریع پرایمرهای PCR قبلاً انتخاب شده برای تعامل ساختارهای پرایمر - دایمر و سنجاق - سری در الیگومرهای DNA کوتاه (کمتر از 30 نوکلئوتید) توسعه داده شده است. AutoDimer در ابتدا برای کمک به توسعه روشهای «PCR چندگانه» (Multiplex - PCR) برای بررسی نشانگرهای STR و SNP برای اهداف پزشکی قانونی ایجاد شد.

Oligo چیست و چه کاربردی دارد؟
OLIGO یکی از نرم افزارهای بیوانفورماتیک تجزیه و تحلیل پرایمر و یک ابزار ضروری برای طراحی و تجزیه و تحلیل توالی و پرایمرهای PCR، ژنهای مصنوعی و انواع پروبها از جمله siRNA و تنظیم کنندههای مولکولی است. بر اساس به روزترین دادههای ترمودینامیکی نزدیکترین همسایه، الگوریتمهای جستجوی Oligo پرایمرهای بهینهای برای PCR، از جمله TaqMan ، پرایمرهای چندگانه، اجماعی یا انحطاط یافته، پیدا میکنند. پردازش دستهای فایلهای متعدد توسط این نرم افزار بیوانفورماتیک امکان پذیر است. همچنین یک ابزار ارزشمند برای «جهش زایی به کار رفته در محل» (site directed mutagenesis) است.

برای هر تک پرایمر یا جفت پرایمر، پنجرههای مختلف تجزیه و تحلیل Oligo تعداد زیادی اطلاعات مفید مانند ساختار ثانویه DNA و RNA، تشکیل دایمر، پرایمینگ غلط و همولوژی، ثبات داخلی، ترکیب و خواص فیزیکی را نشان میدهد. با استفاده از Oligo میتوانید فریمهای باز خواندن را تا وزن مولکولی پیش بینی شده و pKa پروتئینها تجزیه و تحلیل کرده و محلهای آنزیم محدود کننده را نه تنها در DNA بلکه در پروتئینهای معکوس نیز جستجو کنید. اولین نسخه نرم افزار در سال 1989 در بازار ظاهر شد. این برنامه چندین تغییر را انجام داد و آخرین مورد، تغییر از نسخه 6 به 7، جامع ترین آن بود. Oligo 7 میتواند بطور خودکار پرایمرهای چندگانه را انتخاب کرده، فایلهای توالی را در حالتهای دستهای پردازش کند، بطور خودکار پرایمرهای PCR را طراحی میکند تا چندین ناحیه DNA را در یک جستجو بپوشاند و به طور خودکار مجموعه پرایمر/پروب را برای Real - time PCR پیدا میکند یا مجموعه پرایمرهای تو در تو را پیدا میکند.
پروتکلهای جستجوی Oligo (سیستم امتیاز دهی) ممکن است به طور جزئی سفارشی شوند، بنابراین میتوانید نتایج را با توجه به نیازهای خاص خود بهینه کنید. Oligo بر روی مکینتاش و ویندوز اجرا میشود. عملکرد جامع OLIGO با چندین برنامه طراحی اولیه پرایمر تجاری کنار هم مطابقت دارد، بنابراین میتوانید پروبهایی را برای تجزیه و تحلیل توان بالا طراحی کنید، آنها را چندتایی کنید یا بر روی مجموعهای از پرایمر/پروبها تمرکز کنید و آن را برای یک برنامه جدید که هنوز منتشر نشده است بهینه کنید.

IGV چیست و چه کاربردی دارد؟
Integrative Genomics Viewer یا IGV یکی از نرم افزارهای بیوانفورماتیک تعاملی با کارایی بالا، استفاده آسان و راحت برای کاوش بصری دادههای ژنومی است. این برنامه از ادغام انعطاف پذیر انواع رایج دادهها و فرادادههای ژنومی، ایجاد شده توسط محقق یا در دسترس عموم، بارگیری شده از منابع محلی یا ابری پشتیبانی میکند. IGV در اشکال مختلفی وجود دارد، از جمله IGV اصلی که یک برنامه دسکتاپ جاوا است، IGV - Web که یک برنامه بر پایه وب بوده و igv.js که یک جزء جاوا اسکریپت است که میتواند در صفحات وب جاسازی شود.
نرم افزار Chimera چیست؟
UCSF Chimera یک برنامه برای تجسم و تحلیل تعاملی ساختارهای مولکولی و دادههای مربوطه، از جمله نقشههای چگالی، مسیرها و ترازهای توالی است. کایمرا یکی از نرم افزارهای بیوانفورماتیک است که برای استفاده غیر تجاری به صورت رایگان در دسترس است. ChimeraX نسبت به Chimera عملکرد بسیار بهتر برای کار با ساختارهای بزرگ و سایر مزایای عمده و ویژگیهای کاملاً جدیدی دارد. با استفاده از این نرم افزار میتوانید تصاویر و فیلمهای با کیفیت بالا ایجاد کنید. Chimera شامل مستندات کامل است و میتوان آن را به صورت رایگان برای استفاده غیر تجاری بارگیری کرد. Chimera توسط RBVI یا منبع برای محاسبات زیستی، تجسم و انفورماتیک در دانشگاه کالیفرنیا، سان فرانسیسکو توسعه یافته است. برخی از مهمترین ویژگیهای این نرم افزار در ادامه بیان شده اند.
- شناسایی خودکار اتم
- افزودن هیدروژن و تعیین بار جزئی
- اندازه گیری فواصل، زوایا، مساحت سطح و حجم
- ایجاد آسان ویژگیهای سفارشی با ورودیهای ساده فایل متنی
- بسیاری از فرمتها خوانده میشوند، PDB و Mol2 نوشته میشوند.
- گرفتن تصاویر با وضوح بالا
- جلوههای بصری شامل اعلان عمق، سایههای تعاملی، لبههای محیط مرئی، پسزمینههای چند رنگ
- نمایش مولکولی استاندارد
- برچسب گذاری با متن، نمادها، فلشها، کلیدهای رنگی
- ساختارهای مختلف را میتوان متفاوت و در هر زاویه برش داد
- رابط گرافیکی ساده برای ایجاد تعاملی فیلم
- تنظیم آستانه تعاملی، سطوح چندگانه (مش یا جامد)، رندرهای شفاف
- مطابقت مختصات اتمی با نقشهها و نقشهها با هم
- دارا بودن ابزارهای زیادی برای تقسیم بندی و ویرایش نقشهها
- قابلیت خواندن، نوشتن و ویرایش بسیاری از قالبهای هم ترازی توالی
- امکان جستجو BLAST پروتئین از طریق سرویس وب
- چندین روش برای محاسبه حفاظت و نمایش مقادیر در ساختارهای مرتبط

SnapGene چیست و چه کاربردی دارد؟
SnapGene یکی از نرم افزارهای بیوانفورماتیکی و روشی آسان و مطمئن را برای برنامه ریزی، تجسم و مستند سازی روشهای زیست شناسی مولکولی روزمره امکان پذیر میکند. این نرم افزار با رابط بصری، تجسم توالی DNA، حاشیه نویسی توالی، ویرایش توالی، شبیه سازی و تجسم پروتئین روشهای رایج شبیه سازی را امکان پذیر میکند. این نرم افزار همچنین مستندسازی و به اشتراک گذاری دادهها را امکان پذیر میکند. ویژگیهای کلیدی این نرم افزار بیوانفورماتیکی شامل موارد زیر هستند:

- SnapGene تجسم و شبیه سازی دستکاریهای DNA شما را آسان میکند و خطاها را قبل از وقوع به شما هشدار میدهد.
- هرگونه دستکاری DNA در SnapGene به طور خودکار ثبت میشود، بنابراین میتوانید دقیقاً آنچه را انجام داده اید ببینید و توالیهای سازههای اجدادی را بازیابی کنید.
- فایلهای nna SnapGene را میتوان توسط SnapGene Viewer رایگان بین پلتفرمها باز کرد و به شما امکان میدهد نقشهها و توالیهای حاشیه نویسی شده فراوانی را با همکاران به اشتراک بگذارید.
- SnapGene به طور خودکار یک رکورد از هر روش ویرایش و شبیه سازی دنباله ایجاد میکند، بنابراین شما حتی پس از خروج یکی از اعضای آزمایشگاه، نحوه ساخت یک سازه را از دست نخواهید داد.
- SnapGene از تعداد زیادی فرمت فایل پشتیبانی میکند در نتیجه، دانشمندان میتوانند بدون از دست دادن دادهها به طور کامل به SnapGene روی آورند یا میتوانند بدون درگیری به استفاده از نرم افزار قدیمی همراه با SnapGene ادامه دهند.

ابزارهای بررسی همولوژی و شباهت در بیوانفورماتیک
توالیهای همولوگ توالیهایی هستند که با واگرایی از یک جد مشترک مرتبط هستند. بنابراین میزان شباهت بین دو توالی را میتوان اندازه گیری کرد در حالی که همسانی آنها موردی است که یا نادرست است. از این مجموعه ابزارها میتوان برای شناسایی شباهتهای توالیهای جستجوی جدید با ساختار و عملکرد ناشناخته و توالی پایگاه داده که ساختار و عملکرد آنها روشن شده است، استفاده کرد.
BLAST چیست و چه کاربردی در ژنتیک دارد؟
بلاست یا Basic Local Alignment Search Tool در دسته ابزارهای همسان سازی و بررسی شباهت قرار میگیرد. این ابزار مجموعهای از برنامههای جستجو است که برای پلتفرم Windows طراحی شده است و از آن برای جستجوهای شباهت سریع بدون توجه به اینکه پروتکل برای پروتئین است یا DNA میتوانید انجام دهید. مقایسه توالیهای نوکلئوتیدی در پایگاه داده را نیز میتوان با استفاده از این ابزار انجام داد. همچنین میتوان از طریق این ابزار آنلاین یک پایگاه داده پروتئین را برای یافتن مطابقت با توالی پروتئین مورد نظر جستجو کرد. NCBI همچنین سیستم نوبت دهی جدید را به نام Q BLAST به بلاست اضافه کرده است که به کاربران امکان میدهد نتایج را به دلخواه خود بازیابی کرده و نتایج خود را چندین بار با گزینههای قالببندی مختلف اجرا کنند. بسته به نوع توالیهای مورد مقایسه، برنامههای مختلفی در BLAST وجود دارد که شامل موارد زیر هستند:

- blastp توالی پرس و جو آمینو اسید را با پایگاه داده توالی پروتئین مقایسه میکند.
- blastn توالی پرس و جو نوکلئوتیدی را با پایگاه داده توالی نوکلئوتیدی مقایسه میکند.
- blastx یک توالی پرس و جو نوکلئوتیدی ترجمه شده در تمام فریمهای خواندن را با پایگاه داده توالی پروتئین مقایسه میکند.
- tblastn یک توالی پرس و جو پروتئین را با پایگاه داده توالی نوکلئوتیدی مقایسه میکند که به صورت پویا در تمام فریمهای خواندن ترجمه شده است.
- tblastx ترجمههای شش فریم یک دنباله نوکلئوتیدی را با ترجمههای شش فریم پایگاه داده توالی نوکلئوتیدی مقایسه میکند.

FASTA چیست؟
FAST homology search دنبالههای ll برنامه ترازبندی توالیهای پروتئینی که توسط پیرسین و لیپمن در سال 1988 ایجاد شد. این برنامه یکی از بسیاری از الگوریتمهای ابتکاری است که برای تسریع مقایسه توالیها پیشنهاد شده است. ایده اصلی این است که یک مرحله پیش نمایش سریع برای قرار دادن بخشهای بسیار منطبق بین دو دنباله اضافه کنید و سپس این بخشهای منطبق را با استفاده از الگوریتمهای دقیقتر مانند Smith - Waterman به ترازهای محلی گسترش دهید. FASTA میتواند هنگام شناسایی مناطق طولانی با شباهت کم، به ویژه برای توالیهای بسیار واگرا، بسیار ویژه عمل کند. همچنین میتوانید با استفاده از برنامههای FASTA شباهت توالی را در پایگاههای داده نوکلئوتیدی یا پایگاههای داده پروتئوم/ژنوم کامل انجام دهید.
ابزارهای مفید دیگری نیز برای بررسی همولوژی و شباهت توالیها وجود دارند مانند جستجوی توالی ENA که به شما امکان میدهد تا همه توالیهای نوکلئوتیدی موجود در بایگانی نوکلئوتیدهای اروپا (ENA) را جستجو کنید. SSEARCH/GGSEARCH/GLSEARCH با استفاده از برنامههای FASTA و SSEARCH، شباهت توالی را در پایگاههای داده پروتئین ارائه میدهد. SSEARCH یک جستجوی دقیق Smith - Waterman برای یافتن شباهت بین توالی پرس و جو و پایگاه داده انجام میدهد. علاوه بر این موارد ابزار GLSEARCH توالی پروتئین یا DNA را با پایگاه داده توالی مقایسه میکند.

نرم افزار MEGA چیست و چه کاربردی دارد؟
نرم افزار تجزیه و تحلیل ژنتیک تکاملی مولکولی (MEGA)، که در حال حاضر دارای امکاناتی برای ایجاد ترازهای توالی، استنباط تاریخ فیلوژنتیک و انجام تجزیه و تحلیل تکاملی مولکولی است. در نسخه 6 MEGA اکنون استنتاج درختان زمانی را امکان پذیر میکند، زیرا روش RelTime را برای برآورد زمان واگرایی برای همه نقاط انشعاب در فیلوژنی پیاده سازی میکند. یک امکان جدید Timetree در MEGA6 با ارائه یک رابط کاربری گرافیکی (GUI) برای مشخص کردن قدم به قدم محدودیتهای فیلوژنی و کالیبراسیون، این استنباط درخت را تسهیل میکند. این نسخه همچنین شامل الگوریتمهای پیشرفتهای برای جستجوی درختان مطلوب تحت معیارهای تکاملی است و مدیریت حافظه پیشرفتهتری را پیاده سازی میکند که میتواند اندازه مجموعه دادههای توالی را که MEGA برای آنها قابل استفاده است، دو برابر کند. هر دو نسخه GUI و نسخه قابل فرمان دادن MEGA6 را میتوانید از www.megasoftware.net به صورت رایگان دانلود کنید.

نرم افزارهای بیوانفورماتیک تجزیه و تحلیل توالی
این مجموعه از ابزارها به شما امکان میدهند تجزیه و تحلیل دقیقتر و بهتری را در مورد توالی مورد نظر خود از جمله تجزیه و تحلیل تکاملی، شناسایی جهشها، مناطق هیدروپاتی، جزایر CpG و سوگیریهای ترکیبی انجام دهید. شناسایی این موارد و سایر خواص بیولوژیکی همگی سرنخهایی هستند که به جستجو کمک میکند تا عملکرد خاص توالی شما را روشن کنند. به عنوان مثال در بیوانفورماتیک از «هم تراز کردن» (Align) توالیها برای مقایسه 2 توالی که تمام طول هر دو توالی را پوشش میدهد استفاده میشود.

در بیوانفورماتیک، تراز کردن توالی راهی است برای ترتیب دادن توالیهای DNA ، RNA یا پروتئین برای شناسایی مناطق مشابه که ممکن است در نتیجه روابط عملکردی، ساختاری یا تکاملی بین توالیها باشد. توالیهای تراز شده نوکلئوتید یا اسیدهای آمینه به طور معمول به عنوان ردیف در داخل یک ماتریکس نشان داده میشوند. فاصله بین مونومرها ایجاد میشود به طوری که کاراکترهای یکسان یا مشابه در ستونهای پیدرپی تراز میشوند. ترازبندی توالیها همچنین برای توالیهای غیر زیستی مانند محاسبه هزینه فاصله بین رشتهها به زبان طبیعی یا دادههای مالی استفاده میشود.
نرم افزار Editseq چیست؟
EditSeq شما را قادر میسازد تا بر روی توالی اسید نوکلئیک و پروتئین در همه اندازهها از طیف گستردهای از فرمتهای محبوب کار کنید. همچنین میتوانید از رابط اینترنت یکپارچه برای جستجوی پایگاههای اطلاعاتی NCBI برای یافتن توالیها بر اساس شماره دسترسی، شباهت دنباله از طریق BLAST یا جستجوی کلمات کلیدی از طریق پرس و جو متنی Entrez استفاده کنید. برای راحتی کاربر، EditSeq توالی و حاشیه نویسیها را به سه قسمت قابل ویرایش تفکیک میکند. پیوندهای پویا بین دنباله و حاشیه نویسیهای آن هنگام ویرایش یک دنباله، مختصات ویژگی را به طور خودکار به روز میکند و هنگام کپی و چسباندن توالیها شامل اختصاصیت میشود. قابلیتهای موجود همچنین شامل امکان معکوس کردن مکمل، ترجمه توالی و شناسایی ORF ها میشود. مانند دیگر کاربردهای لیزرژن ، EditSeq توالیهایی با اندازههای مختلف از چند نوکلئوتید گرفته تا ژنوم کامل باکتری را مدیریت میکند.
Dna Block Aligner چیست و چه کاربردی دارد؟
نرم افزار بیوانفورماتیک تراز کننده بلوکهای DNA یا DBA، دو توالی را با این فرض که توالیها دارای تعدادی بلوک خطی حفاظتی هستند که با طول بالقوه زیاد و متنوع DNA در دو توالی از هم جدا شده اند، تراز میکند. هدف این بود که این امر برای مناطق سنتزی غیر کد کننده DNA بیانی بین موش و انسان، به عنوان مثال، مناطق بالادست ژن از موش و انسان یا اینترون محافظت شده از ژن انسان - مرغ بسیار منطقی است. بلوکهای محافظت شده ممکن است یک یا دو شکاف داشته باشند. مدل نهایی یک سیستم، حالت محدود احتمالی (یا جفت HMM) است که دو توالی را تراز میکند.
هر بلوک میتواند یکی از 4 مجموعه پارامترهای مختلف را انتخاب کند که تقریباً با 65،75،85 یا 95 درصد هویت آن حفظ میشود. شکافهای خطی (شکافهایی که در آنها شکاف باز شده یکسان است) در بلوکها با احتمال ثابت 0/05 مدل سازی شده اند و هر بلوک حدود 1 درصد از توالی DNA مورد انتظار است. فرم DBA با ارسال دو توالی مورد سوال کار می کند (در صورت لزوم میتوانید از ویژگی بارگذاری فایل استفاده کنید و سپس ارسال کنید. خروجی ترازبندی ASCII به کاربران بازگردانده میشود.
Mauve چیست؟
Mauve یک سیستم برای ایجاد ترازبندی کارآمد چند ژنومی در حضور رویدادهای تکاملی در مقیاس بزرگ مانند بازآرایی و وارونگی است. هم ترازی چندگانه ژنوم، زمینهای برای تحقیقات در زمینه ژنومیک مقایسهای و مطالعه پویایی تکاملی فراهم میکند. تراز کردن ژنومهای کل یک مسئله اساساً متفاوت از تراز کردن توالیهای کوتاه است. Mauve با این ایده توسعه یافته است که یک همسو کننده ژنوم چندگانه فقط به منابع محاسباتی متوسط نیاز دارد. این روش از تکنیکهای الگوریتمی استفاده میکند که از نظر میزان توالی هم تراز به خوبی مقیاس میشوند. به عنوان مثال، یک جفت ژنوم Y. pestis را میتوان در کمتر از یک دقیقه تراز کرد، در حالی که یک گروه 9 ژنومی واگرا انتروباکتریال را میتوان در چند ساعت تراز کرد.

HMMER چیست و چه کاربردی دارد؟
HMMER برای جستجوی پایگاههای داده توالی برای همولوگهای توالیهای پروتئینی و ایجاد تراز توالی پروتئین استفاده میشود. این روشها را با استفاده از مدلهای احتمالی به نام "مدلهای مخفی مارکوف" (HMMs مشخصات) پیاده سازی میکند. در مقایسه با BLAST FASTA و سایر ترازبندیهای توالی و ابزارهای جستجوی پایگاه داده بر اساس روش نمره گذاری قدیمیتر، هدف HMMER این است که به دلیل قدرت مدلهای ریاضی زیرین، برای تشخیص همولوگهای از راه دور بسیار دقیقتر و مناسبتر باشد.در گذشته ، این قدرت با هزینههای محاسباتی قابل توجهی انجام میشد، اما در پروژه جدید HMMER یعنی HMMER۳ در حال حاضر در اصل با سرعت BLAST انجام میشود.
ORF Finder چیست و چه کاربردی در ژنتیک دارد؟
ORF Finder یا Open Reading Frame Finder یک ابزار تجزیه و تحلیل گرافیکی است که همه «فریمهای خواندن باز» (open reading frames) با حداقل اندازه قابل انتخاب را در توالی کاربر یا در توالی موجود در پایگاه داده پیدا میکند. این ابزار با استفاده از کدهای ژنتیکی استاندارد یا جایگزین، تمام فریمهای خواندن باز را شناسایی میکند. دنباله اسید آمینه استنباط شده میتواند در قالبهای مختلف ذخیره شود و با استفاده از سرور WWW.BLAST در پایگاه داده توالی جستجو شود. ORF Finder باید در آماده سازی ارسالهای کامل و دقیق دنباله مفید باشد. همچنین با نرم افزار ارسال دنباله Sequin بسته بندی شده است.
Genome Workbench چیست و چه کاربردی دارد؟
Genome Workbench میتواند دادههای توالی یابی را به طرق مختلفی از جمله نماهای توالی گرافیکی، نماهای هم تراز مختلف، نماهای درخت فیلوژنتیک و نمای جدولی از دادهها نمایش دهد. همچنین میتواند دادههای خصوصی شما را با دادههای پایگاه دادههای عمومی هم تراز کند، دادههای شما را در زمینه دادههای عمومی نمایش دهد و نتایج BLAST را بازیابی کند. Genome Workbench بر روی NCBI C ++ ToolKit ساخته شده است و از API های چند پلتفرمی برای گرافیک استفاده میکند.

این نرم افزار بیوانفورماتیک روی دستگاه محلی شما اجرا میشود و برای Windows 2000/XP ، Linux ، MacOS X و حالتهای مختلف یونیکس در دسترس است.

CpGPlot/CpGReport/Isochore چیست؟
تشخیص مناطقی از توالیهای ژنومی که غنی از الگوی CpG هستند مهم است زیرا چنین مناطقی در برابر متیلاسیون مقاوم هستند و با ژنهایی که اغلب روشن میشوند مرتبط هستند. مناطق غنی از الگوی CpG به عنوان جزایر CpG شناخته میشوند. وظیفه نرم افزار cpg - plot ترسیم مناطق غنی از CpG و تهیه گزارش cpg - report یعنی گزارش همه مناطق غنی از CpG است. ژنومهای هستهای مهره داران موزاییک ایزوکورها، نواحی بسیار طولانی DNA هستند که از نظر ترکیب بازها همگن بوده و از نظر ترکیبی با توالیهای کدگذاری که در آنها جاسازی شده است، ارتباط دارند. Isochores را می توان در تعداد کمی از خانوادهها تقسیم کرد که طیف وسیعی از سطوح GC را پوشش میدهند. برنامه isochore محتوای GC را در یک دنباله ترسیم میکند.

EMBOSS چیست؟
EMBOSS (European Molecular Biology Open Software Suite) یک بسته تجزیه و تحلیل نرم افزاری است. این ابزار میتواند با دادهها در طیف وسیعی از فرمتها کار کند و همچنین دادههای تولی را به صورت شفاف از وب بازیابی کند. کتابخانههای گستردهای نیز با این بسته ارائه شده است که به دانشمندان دیگر اجازه میدهد نرم افزار خود را به عنوان منبع باز منتشر کنند. این ابزارها مجموعهای از برنامههای تجزیه و تحلیل توالی را ارائه میدهند و همچنین از همه سیستم عاملهای UNIX پشتیبانی میکند.
ClustalW
این یک ابزار ترازبندی توالی کاملاً خودکار برای توالی DNA و پروتئین است. این بهترین مطابقت را در طول کل توالیهای ورودی، اعم از پروتئین یا اسید نوکلئیک، ایجاد میکند. اطلاعات اولیهای که آنها ارائه میدهند شناسایی مناطق توالیهای حفظ شده است. این ابزار در طراحی آزمایشات برای بررسی و اصلاح عملکرد پروتئینهای خاص، پیش بینی عملکرد و ساختار پروتئینها و در شناسایی اعضای جدید خانواده پروتئین بسیار مفید است.

توالیها را میتوان در طول (ترازبندی کلی) یا فقط در مناطق خاصی (ترازبندی محلی) تراز کرد. این برای ترازبندیهای جفتی و چندگانه صادق است. تنظیمات کلی باید از شکافها استفاده کنند (نشان دهنده درج/حذف) در حالی که ترازبندیهای محلی میتواند از آنها جلوگیری کند، و مناطق را بین شکافها ترازبندی کند.
ClustalW2 یک برنامه کاملاً خودکار برای ترازبندی چندگانه کلی توالیهای DNA و پروتئین است. هم ترازی پیشرونده است و افزونگی توالی را در نظر میگیرد. این برنامه دارای پارامترهای قابل تنظیم با پیش فرضهای منطقی است. ClustalW یک برنامه همگرا سازی چند منظوره جهانی برای DNA یا پروتئینها است. این از نظر بیولوژیکی دارای توالیهای پشت سر هم متعددی از پیامدهای متفاوت است. این ابزار بهترین تطابق را برای توالیهای انتخاب شده محاسبه میکند و آنها را طوری ترسیم میکند که هویتها، شباهتها و تفاوتها قابل درک باشند. روابط تکاملی را میتوان با مشاهده کلادوگرام یا فیلوگرام مشاهده کرد. ترازبندیهای متعدد توالی پروتئین از ابزارهای مهم تعیین توالی هستند.
نرم افزارهای بیوانفورماتیک تجزیه و تحلیل ساختاری
این مجموعه ابزارها به شما امکان میدهد ساختار مورد نظر خود را با ساختار شناخته شده در پایگاههای داده مقایسه کنید. عملکرد هر پروتئین بیشتر مستقیماً نتیجه ساختار آن است با همولوگهای ساختاری که تمایل دارند عملکردها را تقسیم کنند و کمتر به توالی پروتئین ارتباط دارد. تعیین ساختار دو بعدی یا سه بعدی یک پروتئین در مطالعه عملکرد آن بسیار مهم است. ابزارهایی مانند DaliLite یک برنامه برای مقایسه ساختار جفت است که در آن میتوانید ساختار خود (ساختار اول) را با ساختار مرجع (ساختار دوم) مقایسه کنید. برخی دیگر از نرم افزارهای بیوانفورماتیک مهم در تجزیه و تحلیل ساختاری در ادامه توضیح داده شده اند.
RasMol چیست؟
RASMOL یک برنامه کامپیوتری است که برای تجسم گرافیکی مولکولی نوشته شده است و عمدتاً برای به تصویر کشیدن و اکتشاف ساختارهای ماکرومولکولی بیولوژیکی، مانند آنهایی که در بانک اطلاعات پروتئین موجود است، استفاده میشود. این نرم افزار بیوانفورماتیک در ابتدا توسط راجر سایل در اوایل دهه 90 توسعه یافت. از لحاظ تاریخی، این یک ابزار مهم برای زیست شناسان مولکولی بود زیرا برنامه بسیار بهینه شده اجازه میداد تا نرم افزار بر روی رایانههای شخصی (در آن زمان) بسیار قدرتمند اجرا شود. قبل از RasMol، نرم افزار تجسم بر روی ایستگاههای کاری گرافیکی اجرا میشد که به دلیل هزینه آنها، کمتر در دسترس محققان بود. RasMol به یک ابزار مهم آموزشی تبدیل شده است و همچنان به عنوان یک ابزار مهم برای تحقیقات در زیست شناسی ساختاری شناخته میشود. RasMol شامل زبانی برای انتخاب زنجیرههای پروتئینی خاص، یا تغییر رنگ و غیره است.

PyMOL چیست و چه کاربردی در ژنتیک دارد؟
PyMOL یک سیستم تجسم مولکولی منبع باز، تحت حمایت کاربران است که توسط وارن لیفورد دلانو ایجاد شده و توسط یک شرکت نرم افزاری خصوصی به نام DeLano Scientific LLC تجاری سازی شده است پس به ایجاد ابزارهای مفیدی اختصاص داده شده که برای جوامع علمی و آموزشی در دسترس جهانی قرار میگیرد. برای تولید تصاویر سه بعدی با کیفیت بالا استفاده از مولکولهای کوچک و مولکولهای بیولوژیکی مانند پروتئین مناسب است. تقریباً یک چهارم تمام تصاویر منتشر شده از ساختارهای پروتئینی سه بعدی در مقالات علمی با استفاده از PyMOL تهیه شده است. این ابزار بر پایه «کد منفرد» (single code)، با استفاده از OpenGL و Python و مجموعه کوچکی از وابستگیهای خارجی منبع باز از UNIX ، Macintosh و Windows پشتیبانی میکند.
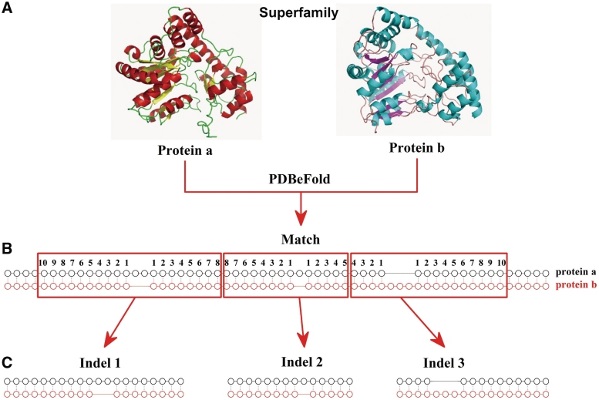
PDBeFold چیست و چه کاربردی در مطالعات ژنتیک دارد؟
PDBeFold (همچنین به عنوان SSM شناخته میشود) یک سرویس تعاملی برای مقایسه ساختارهای پروتئینی به صورت سه بعدی است. این سرویس خدمات زیر را به کاربران خود ارائه میدهد:
- مقایسه زوجی و ترازبندی سه بعدی ساختارهای پروتئینی
- مقایسه چندگانه و ترازبندی سه بعدی ساختارهای پروتئینی
- بررسی ساختار پروتئینی برای شباهت با کل بایگانی PDB یا بایگانی SCOP
- بهترین ترازبندی Cα ساختارهای مقایسه شده
- بارگیری و تجسم سازههای برتر با استفاده از Rasmol (سیستم عاملهای Unix/Linux)، Rastop (ماشینهای MS Windows) و Jmol (بیننده جاوا مستقل از پلت فرم سرور)
- امکان پیوند نتایج به سایر خدمات - PDBeMotif ، SCOP ، GeneCensus ، FSSP ، CATH ، PDBSum ، UniProt.

VMD چیست؟
VMD یا Visual Molecular Dynamics یک برنامه کامپیوتری تجسم و مدل سازی مولکولی است. VMD عمدتا ابزاری برای مشاهده و تجزیه و تحلیل نتایج شبیه سازی دینامیک مولکولی است. همچنین شامل ابزارهایی برای کار با دادههای حجمی، دادههای توالی و اشیاء گرافیکی دلخواه است. صحنههای مولکولی را میتوان به ابزارهای رندر خارجی مانندPOV-Ray ، RenderMan ، Tachyon ، Virtual Reality Modeling Language (VRML) و بسیاری دیگر منتقل کرد. کاربران میتوانند اسکریپتهای Tcl و Python خود را در VMD اجرا کنند زیرا شامل مترجمان تعبیه شده Tcl و Python است. VMD بر روی یونیکس، Apple Mac macOS و Microsoft Windows اجرا میشود.

VMD تحت مجوز مخصوص توزیع در دسترس کاربران غیرتجاری است که هم استفاده از برنامه و هم اصلاح کد منبع آن را بدون هیچ گونه هزینهای مجاز میداند. VMD میتواند از طریق Tcl/Tk با برنامههای دیگر ارتباط برقرار کند. این ارتباط امکان توسعه چندین پلاگین خارجی را فراهم میکند که با VMD کار میکند. این افزونهها مجموعهای از ویژگیها و ابزارهای VMD را افزایش میدهد و آن را به یکی از پرکاربردترین نرم افزارها در شیمی محاسباتی، زیست شناسی و بیوشیمی تبدیل میکند.
Swiss-PdbViewer چیست و چه کاربردی دارد؟
Swiss-PdbViewer (معروف به DeepView) یک برنامه کاربردی است که یک رابط کاربرپسند را برای تجزیه و تحلیل چندین پروتئین به طور همزمان فراهم میکند. پروتئینها را میتوان به منظور استنباط هم ترازیهای ساختاری و مقایسه محلهای فعال آنها یا سایر قسمتهای مربوطه روی هم قرار داد. جهشهای اسید آمینه، پیوندهای هیدروژنی، زوایا و فاصله بین اتمها به دلیل گرافیک بصری و رابط منو به راحتی قابل دستیابی است. Swiss-PdbViewer به SWISS-MODEL، یک سرور مدل سازی خودکار شبیه سازی شده وابسته است که در موسسه بیوانفورماتیک سوئیس (SIB) در گروه سازههای بیوانفورماتیک ساختاری، Biozentrum در شهر بازل ایجاد شده است.
استفاده از این دو برنامه میزان کار لازم برای تولید مدلها را تا حد زیادی کاهش میدهد، زیرا ممکن است یک توالی اولیه پروتئین را روی یک قالب سه بعدی قرار دهد و قبل از ارسال درخواست برای ایجاد حلقههای گمشده و اصلاح بسته بندی، بازخورد فوری از نحوه پذیرش پروتئین قرار گرفته شده توسط ساختار مرجع قبل دریافت کنید. Swiss-PdbViewer همچنین میتواند نقشههای چگالی الکترون را بخواند و ابزارهای مختلفی را برای ایجاد چگالی در اختیار شما قرار میدهد. سرانجام، به عنوان یک امتیاز ویژه، میتواند صحنههای POV-Ray را از نمای فعلی ایجاد کرده تا تصاویری خیره کننده با کیفیت ردیابی اشعه ایجاد کند.

PDBeMotif
PDBeMotif یک ابزار جستجوی بسیار سریع و قدرتمند است که با ترکیب توالی پروتئین، ساختار شیمیایی و دادههای سه بعدی در یک جستجو، اکتشاف در بانک اطلاعات پروتئینی (PDB) را تسهیل میکند. در حال حاضر، این تنها ابزاری است که این نوع ادغام را با چنین سرعتی ارائه میدهد. PDBeMotif را میتوان برای بررسی ویژگیهای محل اتصال پروتئینهای واحد یا کلاس پروتئینها مانند Kinases و ویژگیهای ساختاری محفوظ محیطهای نزدیک آنها در یک گونه یا در گونههای مختلف مورد استفاده قرار داد. به عنوان مثال این ابزار میتواند یک حلقه فعال سازی محافظت شده رایج برای پروتئین کینازها را برجسته کند که در تنظیم فعالیت مهم است و به ترتیب با موتیفهای DFG و APE در ابتدا و انتهای حلقه مشخص شده است. پیش بینی تأثیر تغییرات روی مولکولهای کوچک که به سایتهای فعال یا تنظیم کننده پروتئینها متصل میشوند بر اثربخشی آنها میتواند بر اساس نتیجه کار تحلیلی انجام شده با استفاده از PDBeMotif باشد.
میتوان این نرم افزار بیوانفورماتیک را به تمام سیستم عاملهای اصلی مانند MS Windows ، LINUX ، Apple Mac و Solaris منتقل کرد زیرا در جاوا نوشته شده است و از اوراکل و سرور پایگاه داده منبع رایگان PostGreSQL استفاده میکند. PDBeMotif را میتوان بصورت آنلاین مورد استفاده قرار داد یا بارگیری و نصب محلی کرد که در آن فایلهای PDB عمومی و خصوصی (از جمله کتابخانههای ساختارهای سه بعدی مشتق از نظریه) بارگیری و تجزیه و تحلیل شوند. همچنین قابلیت بارگذاری حاشیه نویسی سایت پروتئینی، خانوادهها و دامنههای سایت پروتئین از سرورهای Distributed Annotation System (DAS) وجود دارد.

Jmol چیست و چه کاربردی دارد؟
Jmol یک نمایشگر جاوا منبع باز برای ساختارهای شیمیایی به صورت سه بعدی است. Jmol یک نمایش سه بعدی از یک مولکول را بر میگرداند که ممکن است به عنوان یک ابزار آموزشی یا برای تحقیقات در شیمی و بیوشیمی استفاده شود. این نرم افزار رایگان و منبع باز بوده که به زبان جاوا نوشته شده است و بنابراین بر روی سیستمهایWindows ، Mac OS X ، Linux و Unix اجرا میشود. یک برنامه مستقل و یک کیت ابزار توسعه وجود دارد که میتواند در سایر برنامههای جاوا ادغام شود.
قابل توجه ترین ویژگی این برنامه کاربردی این است که میتواند برای نمایش مولکولها در انواع مختلف مسیرهای متابولیکی در صفحات وب ادغام شود و از آنها استفاده کند. به عنوان مثال، مولکولها را میتوان به عنوان مدلهای توپ و میله، مدلهای فضا پر کن، مدلهای روبان و غیره نمایش داد. Jmol طیف وسیعی از فرمتهای فایل مولکولی، از جمله بانک اطلاعات پروتئین (pdb)، فایل اطلاعات بلورشناسی (cif)، MDL Molfile (mol) و زبان نشانه گذاری شیمیایی (CML) را پشتیبانی میکند.
نرم افزارهای بیوانفورماتیک تجزیه و تحلیل عملکرد پروتئین
این گروه از برنامهها به شما امکان میدهد ترتیب پروتئین خود را با پایگاه دادههای پروتئینی ثانویه (یا مشتق شده) مقایسه کنید که حاوی اطلاعاتی در مورد موتیفها، رفتار پروتئین و دامنههای پروتئینی است. مراحل بسیار مهم در استفاده از این پایگاه دادههای الگوهای مختلف وجود دارند که به شما امکان میدهند عملکرد بیوشیمیایی پروتئین مورد نظر خود را تقریب بزنید. FingerPRINTScan یک نرم افزار بیوانفورماتیک است که جستجوی پروتکل FingerPRINTScan با دنبالهای از پروتئین برای شناسایی نزدیکترین اثر انگشتی دنباله PRINTS در دنباله پروتئین. تجزیه و تحلیل عملکرد عبارت است از شناسایی و نگاشت تمام عناصر عملکردی (اعم از کدنویسی و غیر کدنویسی) در یک ژنوم. برخی از این ابزارهای بیوانفورماتیکی در زیر شرح داده شده اند.
Molegro چیست؟
Molegro Virtual Docker یک برنامه شبیه سازی اتصال پروتئین - لیگاند است که به ما امکان میدهد شبیه سازی اتصال را در یک بسته محاسباتی کاملاً یکپارچه انجام دهیم. MVD با موفقیت روی صدها پروتئین مختلف با عملکرد docking مشابه سایر برنامههای اتصال مانند AutoDock4 و AutoDock Vina اعمال شده است. Molegro Virtual Docker داکینگ با کیفیت بالا را بر اساس یک تکنیک بهینه سازی جدید همراه با تجربه رابط کاربر با تمرکز بر قابلیت استفاده و بهره وری ارائه میدهد.


برنامه MVD دارای چهار الگوریتم جستجو و چهار تابع نمره دهی بومی است و نشان داده شده است که Docker مجازی Molegro نسبت به سایر محصولات داکینگ پیشرفته دقت اتصال بیشتری دارد. با توجه به حضور یا عدم حضور مولکولهای آب در شبیه سازیهای داکینگ، در مجموع 32 پروتکل اتصال داریم. ادغام برنامههای SAnDReS و MVD امکان انجام تجزیه و تحلیل آماری دقیق نتایج اتصال را فراهم میکنند که به قابلیتهای بومی برنامه MVD میافزاید. دو ویژگی اصلی این برنامه شامل موارد زیر هستند:
- دقت اتصال بالا. ثابت شده است که موتور اتصال به درستی حالتهای اتصال را با دقت بالا شناسایی میکند. نشان داده شده است که Molegro Virtual Docker نسبت به شناسایی حالتهای داکینگ صحت بیشتری نسبت به سایر برنامههای اتصال دارد.
- رابط کاربری آسان. امکانات داخلی این نرم افزار، کاربر را قادر میسازد تا به راحتی راه اندازی و اجرای داکینگ را انجام دهد. ابزارهای تجسم و تجزیه و تحلیل پیشرفته برای بررسی فعل و انفعالات لیگاند - گیرنده و تنظیم دقیق راه حلهای اتصال ارائه شده است.
CluSTr چیست و چه کاربردی دارد؟
دو روش برای جستجوی پایگاه داده CluSTr وجود دارد، جستجوی پیشرفته اجازه درخواستهای مربوط به پروتئین را میدهد و جستجوی ساده اجازه میدهد تا شناسه خوشهای در پایگاه داده جستجو شود. برای جستجوی پیشرفته، از دسترسیهای UniProt Knowledgebase (به عنوان مثال Q9Y9L0) ، شناسههای UniProt Knowledgebase (به عنوان مثال TDXH_AERPE) ، دسترسی IPI (به عنوان مثال IPI00745335) ، شناسههای ایزوفرم UniProt (به عنوان مثال P45983-1) یا نام پروتئین (یا قطعه ای) استفاده کنید. برای جستجوی ساده، خوشههای جداگانه را با مشخص کردن هر سه مورد انتخاب کنید.

Inquisitor چیست؟
این نرم افزار بیوانفورماتیک توالی پروتئین شما را بررسی میکند و مشخص میکند که آیا با دنباله ای در Integr8 (فقط پروتئومهای کامل) و پایگاه دانش UniProt مطابقت دارد یا خیر. اگر توالی مشخص نشده باشد، Inquisitor جزئیات نزدیکترین مطابقتها را به توالی شما باز می گرداند و همچنین تجزیه و تحلیل توالی دقیق ارائه شده را فراهم میکند. Inquisitor از FASTA برای یافتن مطابقتهای نامشخص و از InterProScan برای تجزیه و تحلیل توالیها استفاده میکند. این نرم افزار از طریق یک گزارش وضعیت شما را از روند تجزیه و تحلیل مطلع میکند.
AutoDock چیست؟
AutoDock یک نرم افزار شبیه سازی مدل سازی مولکولی است و یکی از نرم افزارهای بیوانفورماتیک بوده که به ویژه برای اتصال پروتئین - لیگاند موثر است. AutoDock 4 تحت مجوز عمومی GNU در دسترس است. AutoDock 4 در واقع از دو برنامه اصلی تشکیل شده است شامل autodock که متصل شدن لیگاند به مجموعهای از شبکهها که پروتئین مورد نظر را توصیف میکند را انجام میدهد و autogrid که این شبکهها را از قبل محاسبه میکند.

علاوه بر استفاده از آنها برای اتصال، شبکههای میل اتمها را میتوان تجسم کرد. برای مثال، این میتواند به پژوهشگران شیمی آلی سنتزی کمک کند که اتصال دهندههای بهتری را طراحی کنند.

AutoDock Vina جانشین AutoDock است که از نظر دقت و عملکرد به میزان قابل توجهی بهبود یافته است. هر دو AutoDock و Vina در حال حاضر توسط Scripps Research، به ویژه مرکز بیولوژی ساختاری محاسباتی (CCSB) به سرپرستی دکتر آرتور جی اولسون نگهداری میشوند. AutoDock روی لینوکس، Mac OS X ، SGI IRIX و Microsoft Windows اجرا میشود. کامپایل برنامه در حالت 64 بیتی بومی در مایکروسافت ویندوز امکان عملکرد سریعتر نرم افزار را در حالت شناور فراهم میکند. AutoDock به طور گستردهای مورد استفاده قرار میگیرد و در توسعه اولین مهارکننده اینتگراز HIV-1 تایید شده توسط Merck & Co نقش داشته است. AutoDock شامل دو برنامه اصلی AutoDock برای اتصال لیگاند به مجموعهای از شبکههای توصیف کننده پروتئین هدف و AutoGrid برای پیش محاسبه این شبکهها است. AutoDock دارای کاربردهایی در موارد زیر است:
- کریستالوگرافی اشعه ایکس
- طراحی دارو بر اساس ساختار
- بهینه سازی هدایت مولکولی
- «غربالگری مجازی» (HTS)
- طراحی کتابخانه ترکیبی
- اتصال پروتئین - پروتئین
- مطالعات مکانیسم شیمیایی
AutoDock یکی از نرم افزارهای بیوانفورماتیک است که به طور گسترده مورد استفاده قرار گرفته است و نمونههای زیادی از کاربرد موفق آن در مقالات وجود دارد. این نرم افزار بسیار سریع است، پیش بینیهای با کیفیت بالا از تطابق لیگاند و همبستگی خوبی بین ثابتهای مهاری پیش بینی شده و موارد تجربی را ارائه میدهد. همچنین نشان داده شده است که AutoDock در داکینگ کور، که محل اتصال آن مشخص نیست، مفید است. به علاوه، AutoDock یک نرم افزار رایگان است و نسخه 4 تحت مجوز GNU General Public توزیع میشود که به دست آوردن آن نیز آسان است.
InterProScan چیست و چه کاربردی دارد؟
فرم InterProScan به شما امکان میدهد دنباله خود را در برابر InterPro پرس و جو کنید. این پایگاه داده و ابزار تشخیصی یکپارچه است که از روشهای مختلف و میزان متفاوتی از اطلاعات بیولوژیکی در مورد پروتئینهای دارای ویژگی خوب برای به دست آوردن امضای پروتئین استفاده میکند. InterProScan تعدادی پایگاه داده (که به عنوان پایگاه داده اعضا معرفی میشوند) مانند ProDom ، الگوهای PROSITE ، پروفایلهای PROSITE و HAMAP ، PRINTS ، PANTHER ، PIRSF ، Pfam ، SMART ، TIGRFAMs ، Gene3D و SUPERFAMILY را ترکیب میکند.
نرم افزار بیوانفورماتیک GLIMMER
GLIMMER سیستمی برای یافتن ژنها در DNA میکروبی، به ویژه ژنوم باکتریها، آرکئاها و ویروسها است. GLIMMER (Gene Locator and Interpolated Markov Modeler) از مدلهای مارکوف درون یابی شده برای شناسایی مناطق کدگذاری شده استفاده میکند.
نرم افزار بیوانفورماتیک Proteax چیست؟
مجموعه نرم افزاری پروتکس کار با پروتئینهای اصلاح شده و مشتقات پروتئین را آسان میکند. از پروتکس با پایگاه دادههای Microsoft Excel یا Oracle برای ثبت و تجزیه و تحلیل ساختار پروتئین استفاده میشود. موتور Proteax که از نظر شیمیایی دارای اطلاعات زیادی است، محقق را قادر میسازد تا با توالیهای پروتئینی تغییر یافته و شیمیایی کار کند. بنابراین دادههای پروتئینی را میتوان مستقیماً به ابزارهای بیوانفورماتیک، ابزار شیمی و ابزارهای طیف سنجی جرمی منتقل کرد. کارتریج پروتکس مستقیماً از پایگاههای داده Oracle بدون محدودیت در نحوه مدل سازی دادهها پشتیبانی میکند. استفاده از پروتکس در پایگاههای داده PostgreSQL نیز یک گزینه است.
دادههای آفلاین و محلی را میتوان در یک محیط صفحه گسترده آشنا پردازش کرد. Protax برای افزونههای صفحه گسترده در Microsoft Excel و OpenOffice.org Calc کار میکند و عملکرد کامل مجموعه ابزار Proteax را دقیقاً در صفحه گسترده مورد علاقه شما ارائه میدهد. پردازش انبوه مجموعه دادههای فایل مسطح برای پروتکس دسکتاپ مناسب است که میتواند توسط همه زبانهای برنامه نویسی رایج اسکریپت شود.

RADAR چیست و چه کاربردی دارد؟
مخفف RADAR عبارت Rapid Automatic Detection and Alignment of Repeats به معنی تشخیص سریع خودکار و تنظیم تکرار پروتئین در دنبالههای پروتئینی است. بسیاری از پروتئینهای بزرگ با تکثیر داخلی تکامل یافته اند و بسیاری از تکرارهای داخلی مربوط به واحدهای عملکردی و ساختاری است. رادار از یک الگوریتم خودکار برای تقسیم بندی توالی مورد نظر به تکرارها استفاده میکند. این ترکیب کوتاه مغرضانه و همچنین تکرارهای تقریبی و معماری تکرار پیچیده را که شامل انواع مختلف تکرارها در توالی مورد نظر شما میشود، شناسایی میکند.
کاربرد زبان های برنامه نویسی در بیوانفورماتیک
زبانهای برنامه نویسی امروزه کاربردهای بسیار وسیعی در بیوانفورماتیک پیدا کرده اند. به عنوان مثال از آنجا که مراکز تحقیقاتی در سراسر جهان از محیط خصوصی تا دانشگاهی پراکنده شده اند و طیف وسیعی از سخت افزارها و سیستم عاملها مورد استفاده قرار میگیرند، جاوا به عنوان یک بازیگر کلیدی در بیوانفورماتیک در حال ظهور است. فن آوریهای شبیه سازی بیولوژیکی مبتنی بر رایانه و PatternHunter Bioinformatics Solutions دو مثال از پذیرش فزاینده جاوا در بیوانفورماتیک هستند. پروژه BioJava به ارائه ابزار جاوا برای پردازش دادههای بیولوژیکی اختصاص دارد که شامل اشیایی برای دستکاری توالیها، برنامه نویسی پویا، تجزیه کنندههای فایل، روالهای آماری ساده و غیره است. علاوه بر آن زبانهای برنامه نویسی دیگری نیز وجود دارند که در بیوانفوراتیک کاربرد دارند و در ادامه به بررسی آنها میپردازیم.
زبان برنامه نویسی R در بیوانفورماتیک
R یکی از زبانهای برنامه نویسی برجسته در علم داده است. به طور گستردهای برای انجام آمار، machine learning، تجسم و تجزیه و تحلیل دادهها استفاده میشود. این یک زبان برنامه نویسی منبع باز است. R یکی از پرکاربردترین و قدرتمندترین زبانهای برنامه نویسی در بیوانفورماتیک است و به ویژه در جایی که ابزارهای آماری مختلفی مورد نیاز است (مانند RNA-Seq ، ژنومیک جمعیت و غیره) و در تولید نمودارها و ارقام با کیفیت انتشار نقش بسیار مهمی دارد.
Perl در بیوانفورماتیک
دستکاری رشته، تطبیق عادی عبارت، تجزیه فایل، تبدیل فرمت داده و غیره کارهای متداول پردازش متن است که در بیوانفورماتیک انجام می شود. زبان Perl در چنین وظایفی برتری دارد و توسط بسیاری از توسعه دهندگان مورد استفاده قرار میگیرد. با این وجود، هیچ ماژول استانداردی که در Perl به طور خاص برای حوزه بیوانفورماتیک طراحی شده باشد وجود ندارد. با این حال، توسعه دهندگان چندین ماژول شخصی خود را برای این منظور طراحی کرده اند که بسیار محبوب شده اند و توسط پروژه BioPerl هماهنگ میشوند. پروژه BioPerl یک انجمن بین المللی توسعه دهندگان ابزار Perl برای بیوانفورماتیک است و منبع آنلاین ماژولها، اسکریپتها و پیوندهای وب را برای توسعه دهندگان نرم افزارهای مبتنی بر Perl ارائه میدهد.

Biopython چیست؟
Biopython مجموعهای از ابزارهای رایگان موجود برای محاسبات بیولوژیکی است که توسط تیم توسعه دهندگان بین المللی در پایتون نوشته شده است. این یک تلاش مشترک توزیع شده برای توسعه کتابخانهها و برنامههای کاربردی پایتون است که نیازهای کارهای فعلی و آینده در بیوانفورماتیک را برطرف میکند. پایتون یک زبان شی گرا، تفسیر شده و انعطاف پذیر است که برای محاسبات علمی رو به توسعه است. یادگیری پایتون آسان است، نحو بسیار واضحی دارد و به راحتی میتوان آن را با ماژولهای نوشته شده در C ، C ++ یا FORTRAN گسترش داد.

اساساً، هدف Biopython این است که با ایجاد ماژولها و کلاسهای باکیفیت و قابل استفاده مجدد، استفاده از Python را برای بیوانفورماتیک تا حد ممکن آسان کند. ویژگیهای Biopython شامل تجزیه کنندهها برای فرمتهای مختلف فایلهای بیوانفورماتیک (BLAST ، Clustalw ، FASTA ، Genbank ، ...) ، دسترسی به خدمات آنلاین (NCBI ، Expasy ، ...) ، رابط برنامههای رایج و نه چندان رایج (Clustalw ، DSSP ، MSMS ...) است. نسخههای اصلی Biopython دارای قابلیتهای زیادی هستند که در ادامه در مورد آنها صحبت کرده ایم.
- توانایی تجزیه فایلهای بیوانفورماتیک در ساختارهای داده قابل استفاده در پایتون، از جمله پشتیبانی از فرمتهای Blast .Clustalw ، FASTA ، GenBank ، PubMed ، ExPASy ، SCOP ، UniGene ، SwissProt
- فایلها در قالبهای پشتیبانی شده را میتوان با ضبط تکرار کرد یا نمایه سازی کرد و از طریق رابط فرهنگنامه این زبان برنامه نویسی به آنها دسترسی پیدا کرد.
- کدگذاری برای همکاری با مقاصد محبوب بیوانفورماتیک آنلاین مانند NCBI و خدمات Blast ، Entrez و PubMed و همچنین EXPASy ورودیهای Swiss - Prot و Prosite و جستجوهای Prosite.
- رابط برنامههای رایج بیوانفورماتیک مانند Blast مستقل از NCBI ، برنامه ترازبندی Clustalw، ابزارهای خط فرمان EMBOSS
- یک کلاس توالی استاندارد که به توالیها، شناسههای توالیها و ویژگیهای توالی میپردازد.
- ابزارهایی برای انجام عملیات متداول در توالیها، مانند ترجمه، رونویسی و محاسبه وزن.
- کدگذاری برای همکاری با ترازبندیها، شامل یک روش استاندارد برای ایجاد و برخورد با ماتریکسهای جایگزین.
- برنامههای مبتنی بر رابط کاربری گرافیکی برای انجام عملیات اصلی، ترجمه، BLASTing و غیره.